



Содержание
Перейти к:
 О. В. Васюкова,
Е. П. Атавина-Ермакова,
Е. Г. Панченко,
Д. А. Копытина,
О. А. Симонова,
В. Ю. Удалова,
И. В. Канивец,
Д. Б. Акимова,
М. Ю. Скоблов,
М. А. Андреева,
Е. В. Нагаева,
Н. Г. Мокрышева
О. В. Васюкова,
Е. П. Атавина-Ермакова,
Е. Г. Панченко,
Д. А. Копытина,
О. А. Симонова,
В. Ю. Удалова,
И. В. Канивец,
Д. Б. Акимова,
М. Ю. Скоблов,
М. А. Андреева,
Е. В. Нагаева,
Н. Г. Мокрышева https://doi.org/10.14341/omet13276
Перейти к:
Обоснование. Болезни геномного импринтинга — это особый класс наследственных заболеваний, обусловленных нарушением моноаллельной экспрессии генов. Некоторые из них тесно связаны с развитием ожирения и метаболических расстройств. Такие синдромы, как Прадера-Вилли, Ангельмана, Шаафа-Янг, Темпл, псевдогипопаратиреоз 1а и 1b типов, иллюстрируют, как дисрегуляция экспрессии импринтированных генов может приводить к нарушению энергетического баланса, гиперфагии, снижению физической активности и аномальному распределению жировой ткани. В настоящее время в структуре раннего и тяжелого ожирения неуклонно возрастает удельный вес случаев, обусловленных именно генетическими причинами.
Цель. Изучить клинические и генетические характеристики синдромальных форм ожирения у детей.
Материалы и методы. Ретроспективное несравнительное исследование. В исследование включены 186 пациентов, обследованных в ГНЦ ФГБУ «НМИЦ эндокринологии имени академика И.И. Дедова» с подозрением на генетические формы ожирения в период с октября 2022 по май 2025 гг.
Результаты. У 12% пациентов (n=22) было подтверждено наличие болезней геномного импринтинга: синдром Прадера-Вилли (n=12), синдром Ангельмана (n=1), синдром Шаафа-Янг (n=1), синдром Темпл (n=1), мультилокусные нарушения импринтинга (MLID) (n=1), псевдогипопаратиреоз 1а типа (n=6). На момент обследования избыточный вес (SDS ИМТ 1,0–1,9 SD) имели 13,6% (n=3), ожирение 1 степени (SDS ИМТ 2,0 2,4 SD) имели 4,6% (n=1), ожирение 3 степени (SDS ИМТ 3,0–3,9 SD) имели 18,2% (n=4), морбидное ожирение (SDS ИМТ≥4,0 SD) — 40,9% (n=9). Избыточный набор массы тела на 1-м году жизни отмечался в 40,9% случаев (n=9), и в 31,8% (n=7) со 2-го года жизни. Полифагия клинически была выявлена в 54,5% случаев (n=12). Наследственность по ожирению была отягощена в 31,8% случаев (n=7). Медиана SDS ИМТ составила 3,71 SD [1,8025; 4,2875]. У 13 из 17 пациентов отмечались осложнения ожирения и избыточной массы тела.
Заключение. В работе представлены генетические и клинические характеристики болезней геномного импринтинга в структуре синдромального ожирения у детей в Российской Федерации.
Васюкова О.В., Атавина-Ермакова Е.П., Панченко Е.Г., Копытина Д.А., Симонова О.А., Удалова В.Ю., Канивец И.В., Акимова Д.Б., Скоблов М.Ю., Андреева М.А., Нагаева Е.В., Мокрышева Н.Г. Болезни геномного импринтинга в структуре синдромального ожирения у детей. Ожирение и метаболизм. 2025;22(3):180-195. https://doi.org/10.14341/omet13276
Vasyukova O.V., Atavina-Ermakova E.P., Panchenko E.G., Kopytina D.A., Simonova O.A., Udalova V.Y., Kanivets I.V., Akimova D.B., Skoblov M.Y., Andreeva M.A., Nagaeva E.V., Mokrysheva N.G. Genomic imprinting disorders in the structure of syndromic obesity in children. Obesity and metabolism. 2025;22(3):180-195. (In Russ.) https://doi.org/10.14341/omet13276
Детское ожирение в настоящее время рассматривается как одна из наиболее значимых медико-социальных проблем. Согласно данным Всемирной организации здравоохранения, во всем мире наблюдается стабильный рост распространенности избыточной массы тела и ожирения среди детей и подростков. Этот феномен приобретает особенности эпидемии и сопряжен с высоким риском развития метаболических, сердечно-сосудистых, эндокринных и психоэмоциональных нарушений уже в раннем возрасте.
Ожирение, возникшее в детском возрасте, как правило, персистирует и во взрослом состоянии, значительно повышая риск преждевременной инвалидизации и смертности. Особую обеспокоенность вызывает тенденция к снижению возраста дебюта заболевания, что коррелирует с глобальными изменениями в образе жизни, включая снижение уровня физической активности, нарушение пищевого поведения и воздействие социальных факторов.
В то же время, несмотря на преобладание конституционально-экзогенных форм ожирения, в структуре раннего и тяжелого ожирения неуклонно возрастает удельный вес случаев, обусловленных генетическими причинами. Это определяет необходимость углубленного изучения патогенеза, своевременной диагностики и персонифицированного подхода к лечению и профилактике детского ожирения.
По данным литературы, генетические формы ожирения составляют от 5 до 10% детского ожирения [65]. Генетическое ожирение, в свою очередь, подразделяется на синдромальное и несиндромальное. Причинами синдромального ожирения могут быть генные, хромосомные мутации и аномалии метилирования в контексте геномного импринтинга.
В диплоидных клетках млекопитающих большинство аутосомных генов одинаково экспрессируются отцовскими и материнскими аллелями, что приводит к биаллельной экспрессии [1]. Однако существует небольшая подгруппа генов, которые демонстрируют моноаллельную экспрессию в зависимости от родительского происхождения аллеля (материнского или отцовского), второй же аллель в данном случае импринтирован (моноаллельно метилирован) и не экспрессируется [1]. Это явление называется геномным импринтингом (ГИ).
ГИ является формой неменделевского наследования, которая контролируется эпигенетическими метками, установленными по-разному в родительской зародышевой линии, без изменения последовательности ДНК. За исключением индивидуальных полиморфизмов последовательностей, которые не связаны со статусом импринтинга, последовательности ДНК двух родительских аллелей импринтированных генов идентичны. Таким образом, для достижения родительской специфической экспрессии гомологичные хромосомы должны быть различимы по определенной эпигенетической метке. Изученные до сих пор импринтированные области генов в основном показывают различия в метилировании ДНК между родительскими аллелями. Последовательность, способствующая этому эпигенетическому контрасту, известна как дифференциально метилированная область (DMR). В импринтированных локусах были описаны два типа DMR — один из них приобретает метилирование во время гаметогенеза (герминальный DMR), а другой становится метилированным после оплодотворения (соматический DMR), что зависит от работы зародышевого DMR [6]. Импринтированные гены, как правило, организованы в кластеры, многие из которых находятся под контролем ключевых цис-действующих локусов, называемых импринтинг-контролирующими областями (ICR), иногда центрами импринтинга (IC) [6]. Основным эпигенетическим модификатором генома является метилирование цитозиновых оснований ДНК, определяющее взаимодействие между ДНК и белками, распознающими модифицированные основания, и регулирующее экспрессию генов через механизм компактизации — декомпактизации хроматина [2].
Окончательное доказательство геномного импринтинга у млекопитающих было получено в ходе серии экспериментов на мышах с трансплантацией пронуклеусов в начале 1980-х годов [3][4]. Дальнейшие исследования сузили регионы, содержащие эффекты родительского происхождения, до кластера генов, а в некоторых случаях и до отдельных генов. В 1991 г. был идентифицирован первый мышиный импринтированный ген — ген рецептора инсулиноподобного фактора роста 2 типа (Igf2r) [5]. В настоящее время известно уже около 100 импринтированных генов, многие из которых оказывают существенное влияние на рост и развитие плода. У человека наиболее хорошо описанные кластеры импринтированных генов находятся на хромосомах 6, 7, 11, 14, 15, 20.
Нарушения моноаллельной экспрессии генов приводят к развитию особого класса наследственных заболеваний человека — болезней геномного импринтинга (БГИ) [2]. Первыми зарегистрированными БГИ у человека были синдром Прадера-Вилли (СПВ) и синдром Ангельмана (СА).
Синдром Ангельмана — это орфанное генетическое заболевание, которое может быть вызвано делецией региона 15q11.2–13 хромосомы 15 материнского происхождения, мутацией гена UBE3A (локус 15q11.2) хромосомы 15 материнского происхождения, однородительской дисомией региона 15q11.2–13 отцовского происхождения, дефектами центра импринтинга [7]. Это классический пример геномного импринтинга, при котором экспрессия геномной области различается в зависимости от родительского происхождения аллеля. Распространенность данного заболевания в популяции оценивается в 1:12 000-1:24 000 человек [50]. Точных статистических данных о количестве больных с синдромом Ангельмана в мире, в том числе в России, нет.
Впервые заболевание описал английский педиатр H. Angelman в 1965 г., который наблюдал 3 детей c умственной отсталостью, двигательными нарушениями и особенностями поведения. H. Angelman назвал их «дети-марионетки» из-за необычной позиции рук и отрывистых движений конечностей.
Согласно консенсусу, основными клиническими признаками СА являются тяжелая задержка развития и умственная отсталость, дефекты речи, атаксия походки и/или тремор конечностей, приступы судорог, а также следующие особенности поведения — частый смех/улыбки, гипервозбудимость, стереотипные движения рук [50]. Ожирение относится к признакам, встречающимся реже, чем у 80% пациентов с СА [51].
Синдром Шаафа–Янг (СШЯ) — это редкое аутосомно-доминантное заболевание, которое характеризуется расстройствами аутистического спектра (РАС), контрактурами суставов и глубокой дисфункцией гипоталамуса. Ожирение на фоне гиперфагии при СШЯ развивается чаще всего уже во взрослом возрасте. СШЯ был впервые описан C.P. Shaaf и соавторами в 2013 г. у 4 неродственных пациентов [49]. На сегодняшний день выявлено более 250 человек с СШЯ, хотя его истинная распространенность до конца не известна и, предположительно, составляет 1 случай на 1 миллион человек [40][22].
Синдром Темпл (СТ) — редкое расстройство импринтинга. Распространенность данного синдрома составляет менее 1 случая на миллион. Первый пациент с СТ был описан И.К. Темпл в 1991 г. [62].
Классические фенотипические признаки СТ включают пренатальную и постнатальную задержку роста, акромикрию, относительную макроцефалию, выступающие лобные бугры, мышечную гипотонию, трудности с кормлением, гонадотропинзависимое преждевременное половое развитие [59][60][61]. СТ также часто сопровождается плацентарной гипоплазией, задержкой психомоторного развития, ожирением и его метаболическими осложнениями, такими как гиперхолестеринемия и сахарный диабет [59][60][61].
Таким образом, некоторые из БГИ тесно связаны с развитием выраженного ожирения и метаболических расстройств. Однако специфической этиопатогенетической терапии для данных состояний в настоящее время нет.
Изучить клинические и генетические характеристики синдромальных форм ожирения у детей.
В исследование включены 186 пациентов. От каждого пациента/представителя пациента было получено письменное информированное согласие. Клинические, лабораторные и инструментальные данные получены из медицинских карт на дату первой госпитализации/обследования, если таких было более одного.
Критерии включения: пол: дети обоих полов (мальчики и девочки). Возраст: от 1 до 215 мес. Диагноз по коду МКБ 10: Е66.8, Q87.1, Q87.8.
Критерии исключения: опухоли хиазмально-селлярной области в анамнезе. Моногенные и полигенные формы ожирения.
Сплошной способ формирования выборки.
Данная часть исследования является одноцентровым ретроспективным несравнительным исследованием. Набор пациентов в группы проводился на основании соответствия критериям включения и при отсутствии критериев исключения.
Протокол исследования содержал клиническое обследование пациентов с подробным сбором наследственного анамнеза, с физикальным осмотром и оценкой фенотипических особенностей, антропометрических показателей (расчет SDS роста, SDS ИМТ проведен с помощью компьютерной программы Auxology 1,0 (Pfizer, США)). Лабораторная диагностика включала исследование уровня гликированного гемоглобина (HbA1c), гемоглобина, эритроцитов, гематокрита, креатинина, глюкозы крови натощак, триглицеридов (ТГ), общего холестерина, липопротеинов низкой плотности (ЛПНП), липопротеинов высокой плотности (ЛПВП), аланинаминотрансферазы (АЛТ), аспартатаминотрансферазы (АСТ), железа сывороточного крови, ферритина. Гормональное исследование включало определение уровня инсулина (ИРИ), тиреотропного гормона (ТТГ), уровня свободного тироксина (свТ4), паратгормона (ПТГ), инсулиноподобного фактора роста — 1 (ИПФР-1), пролактина, кортизола, адренокортикотропного гормона (АКТГ), лютеинизирующего гормона (ЛГ), фолликулостимулирующего гормона (ФСГ), эстрадиола, дегидроэпиандростерон-сульфата (ДГЭА-С). Инструментальная диагностика включала в себя ультразвуковое исследование органов брюшной полости (УЗИ ОБП), щитовидной железы (УЗИ ЩЖ), биоимпедансометрию. Наличие инсулинорезистентности оценивалось на основании индекса Matsuda, определяемого по данным глюкозотолерантного теста (ОГТТ) — значения индекса ниже 2,6 свидетельствуют о наличии инсулинорезистентности [68]. Наличие неалкогольной жировой болезни печени (НАЖБП) оценивалось по ультразвуковому исследованию органов брюшной полости и уровню АЛТ и АСТ — диагностическими ультразвуковыми признаками жирового гепатоза являлись гепатомегалия, неоднородность паренхимы и ослабление ультразвукового сигнала в дистальных отделах печени, обеднение сосудистого рисунка, активность АЛТ и АСТ, превышающая верхнюю границу нормы, установленной для данной лаборатории, у детей с признаками НАЖБП по УЗИ при отсутствии других причин синдрома цитолиза (вирусные, метаболические поражения печени и др.) расценивалась как проявление стеатогепатита [68]. Наличие дислипидемии устанавливалось при наличии двух и более «высоких» и/или «низких» показателей: холестерин ≥5,2 ммоль/л; триглицериды >1,3 (для детей до 10 лет); ≥1,7 (для детей старше 10 лет) ммоль/л; ЛПВП ≤0,9 (мальчики) и ≤1,03 (девочки) ммоль/л; ЛПНП ≥3,0 ммоль/л [68].
Анализ числа копий и статуса метилирования импринтированных районов 6q24.2; 7q32.2; 11p15.5; 14q32.2; 15q11.2; 19q13.43; 20q13.32 методом метил-чувствительной мультиплексной лигазо-зависимой амплификации зондов (МЧ-MLPA) проводили с помощью реактивов “SALSA MLPA Probemix ME034 Multi-locus Imprinting” производителя MRC Holland на образцах ДНК, выделенных из лимфоцитов периферической крови пробанда. Выделение ДНК осуществляли методом фенол-хлороформной экстракции. Концентрацию ДНК измеряли на приборе Qubit 2.0 с использованием реактивов Qubit BR. Интерпретацию результатов фрагментного анализа осуществляли с помощью программы Coffalyser производителя MRC Holland.
Микросателлитный анализ локусов критического района хромосомы 15q11.2 (D15S11, D15S113, D15S128, D15S1021, D15S817, D15S1513, D15S986) для определения однородительской дисомии (ОРД) проводили с использованием ДНК, выделенной из периферической венозной крови пробанда и родителей. Выделение ДНК осуществляли методом фенол-хлороформной экстракции. Концентрацию ДНК измеряли на приборе Qubit 2.0 с использованием реактивов Qubit BR . Интерпретацию результатов фрагментного анализа осуществляли с помощью программы GeneMapper6.
ДНК пациента была проанализирована методом секвенирования нового поколения (2 × 151 п.н. парных прочтений). Библиотека ДНК была обогащена методом селективного захвата, направленного на белок-кодирующие области генов человека.
РНК-анализ гена MAGEL2 проводили на образцах тотальной РНК, выделенной из первичной культуры фибробластов, полученных в сетевом центре коллективного пользования «Всероссийская коллекция биологических образцов наследственных болезней» (МГНЦ). Выделение РНК выполнено с использованием реагента Extract RNA («Эвоген», Россия). Обратную транскрипцию проводили с использованием системы Reverse Transcription System («Диалат», Россия) в соответствии с рекомендациями производителя. Качество полученной кДНК оценивали с помощью количественной ПЦР гена B2M.
Для РНК-анализа варианта NM_019066.5:c.2457T>A гена MAGEL2 проводили амплификацию целевого локуса с использованием следующих праймеров: 5’-CTGGGCTCCGCTAAATCAT-3’ и 5’-TTTGGACCTCCCAGTCACTC-3’. Анализ полученного ПЦР-продукта проводился с помощью таргетного секвенирования нового поколения (NGS).
Библиотеки для NGS подготовлены с помощью набора «SG GM» («Raissol») и отсеквенированы на платформе FASTASeq методом парно-концевых прочтений (2×150 н.п.). Обработка сырых данных секвенирования проведена с помощью специализированного биоинформатического пайплайна на основе открытых инструментов. Алгоритм включает контроль качества прочтений с использованием FastQC v0.12.1, картирование и сортировку прочтений относительно референсного генома человека hg19 с помощью STAR 2.7.11b.
Расчет данных производился с помощью статистического пакета Statistica 8 (StatSoft inc., США), MS Exel 2016 (Microsoft, США). Количественные результаты представлены в виде медианы (Ме) и квартилей [ Q1; Q3], соответствующих 25 и 75 перцентилям.
Проведение исследования одобрено локальным этическим комитетом ГНЦ ФГБУ «НМИЦ эндокринологии имени академика И.И. Дедова» Минздрава России 09.10.2024 г.
В исследование включено 186 пациентов (102 мальчика, 84 девочки). У 22 пациентов (12%) было подтверждено наличие болезней геномного импринтинга: синдром Прадера-Вилли (n=12), синдром Ангельмана (n=1), синдром Шаафа-Янг (n=1), синдром Темпл (n=1), мультилокусные нарушения импринтинга (MLID) (n=1) [67], псевдогипопаратиреоз 1а тип (n=6) (рис. 1) [69].
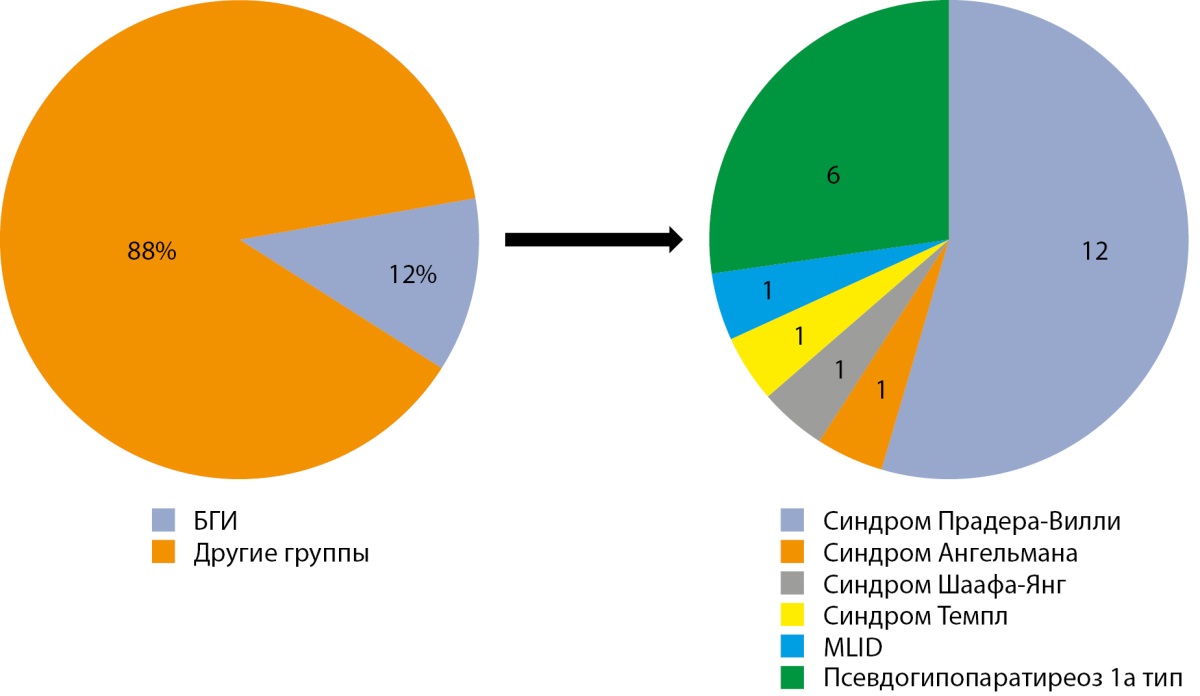
Рисунок 1. Частота БГИ в структуре исследуемой группы пациентов.
Медиана возраста обращения пациентов с БГИ в ГНЦ ФГБУ «НМИЦ эндокринологии имени академика И.И. Дедова» составила 71 месяц [ 29,5; 113,75]. Среди них было 13 мальчиков и 9 девочек.
В неонатальном периоде диффузная мышечная гипотония и слабый сосательный рефлекс отмечались в 63,6% случаев (n=14). Крипторхизм среди мальчиков отмечался в 69,2% случаев (n=9). Эпизоды гипогликемий отмечались в 13,6% случаев (n=3). Задержка развития на первом году жизни отмечалась в 77,3% случаев (n=17).
На момент обследования дефицит массы тела (SDS ИМТ≤-2 SD) имели 13,6% (n=3), нормальный вес (SDS ИМТ -1,9–0,9 SD) имели 9,1% (n=2), избыточный вес (SDS ИМТ 1,0–1,9 SD) имели 13,6% (n=3), ожирение 1 степени (SDS ИМТ 2,0–2,4 SD) имели 4,6% (n=1), ожирение 3 степени (SDS ИМТ 3,0–3,9 SD) имели 18,2% (n=4), морбидное ожирение (SDS ИМТ≥4,0 SD) — 40,9% (n=9). Избыточный набор массы тела на 1 году жизни отмечался в 40,9% случаев (n=9), и в 31,8% (n=7) со 2-го года жизни. Полифагия клинически была выявлена в 54,5% случаев (n=12). Наследственность по ожирению была отягощена в 31,8% случаев (n=7). Медиана веса составила 36,75 кг [ 16,875; 54,175]. Медиана SDS ИМТ составила 3,71 SD [ 1,8025; 4,2875].
Задержку роста (SDS роста≤-2 SD) имели 22,7% (n=5). Медиана SDS роста составила 0,275 SD [ -1,4025; 0,7475].
У 13 из 17 пациентов отмечались осложнения ожирения и избыточной массы тела (рис. 2). Нарушения углеводного обмена были выявлены в 41,2% случаев (n=7): в 23,5% случаев (n=4) имела место инсулинорезистентность, а в 17,7% случаев (n=3) — инсулинорезистентность в сочетании с нарушением толерантности к глюкозе. В 41,2% случаев (n=7) была выявлена дислипидемия. 11 пациентов (64,7%) имели неалкогольную жировую болезнь печени: в 35,3% случаев (n=6) имел место жировой гепатоз, а в 29,4% (n=5) — стеатогепатит. Артериальная гипертензия отмечалась в 11,75% случаев (n=2). Иные этиологические причины состояний были исключены.
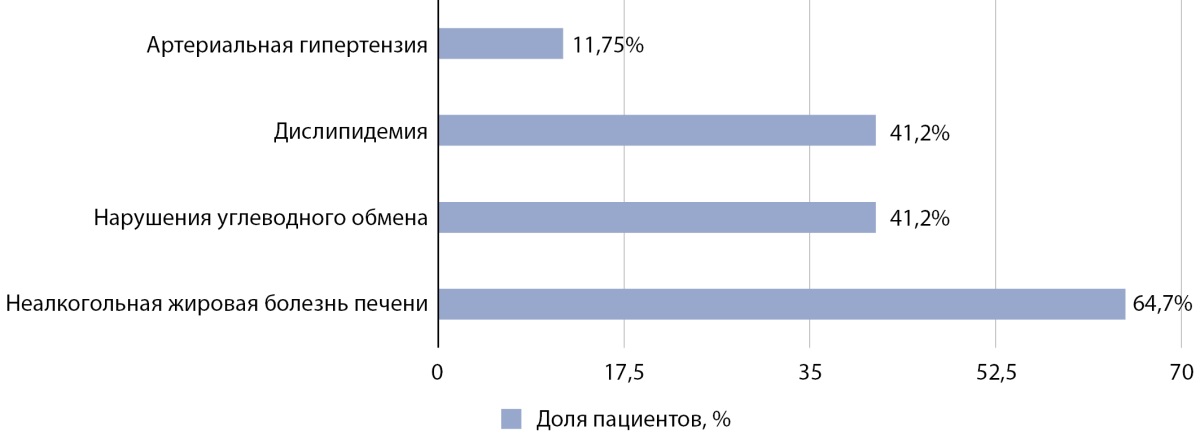
Рисунок 2. Частота осложнений, ассоциированных с ожирением и избыточной массой тела, у пациентов с БГИ.
Сопутствующую эндокринную патологию имели 54,5% пациентов (n=12): в 18,2% случаев (n=4) отмечался вторичный гипотиреоз, в 9,1% случаев (n=2) — сочетание вторичного гипотиреоза с гипогонадотропным гипогонадизмом. В 27,2% случаев (n=6), в рамках мультигормональной резистентности при псевдогипопаратиреозе 1а типа, отмечались гипотиреоз (резистентность к ТТГ), гиперпаратиреоз с гипокальциемией (резистентность к ПТГ) и гиперкальцитонинемия (резистентность к кальцитонину).
Из фенотипических особенностей у пациентов чаще всего отмечались выступающий лоб, долихоцефалия, микрогнатия, готическое небо, миндалевидный разрез глаз, высокая спинка носа, опущенные уголки рта, низкопосаженные уши, акромикрия, диффузная мышечная гипотония, лунообразное лицо, короткая шея, подкожные кальцификаты, брахидактилия, укорочение пальцев за счет IV, V пястных и плюсневых костей.
Учитывая клинический полиморфизм БГИ, приводим описания клинических случаев.
Девочка, 7 лет 9 месяцев. Родилась от 2-й беременности, протекавшей без патологии. Роды на 39-й неделе путем кесарева сечения, асфиксия в родах. При рождении: длина тела — 54 см (SDS 2,54), вес — 3950 г (SDS 1,46).
Наследственный анамнез: брак не близкородственный. Мама — 158 см, 68 кг (ИМТ — 27,2 — избыточная масса тела); папа — 179 см, 64 кг (ИМТ 20,0 — норма). Родная сестра (13 лет) здорова, имеет нормальные весо-ростовые показатели.
С первых месяцев жизни у девочки отмечалась задержка психомоторного развития (голову начала держать с 3-х месяцев, переворачивается с 5-ти месяцев, сидит с 10 месяцев, ходит с поддержкой с 1,5 года), в связи с чем наблюдалась у невролога. С двух лет, по данным ЭЭГ, фиксировалась высокая эпилептиформная активность. С 3,5 года наблюдалась с диагнозом «Резидуально-органическое поражение ЦНС. Эпилептическая генетическая энцефалопатия. Сенсомоторная алалия. Умственная отсталость». В 4,5 года была инициирована противоэпилептическая терапия леветирацетамом и клобазамом.
В 4 года 10 месяцев проведено молекулярно-генетическое исследование (инициировано неврологами) — у девочки и папы найден ранее не описанный, вероятно патогенный вариант в гене SCN1А, однако найденная мутация не соответствовала клинической картине.
Также, помимо неврологических особенностей, со второго месяца жизни у девочки отмечалась избыточная месса тела на фоне полифагии (+2 кг ежемесячно). Со слов мамы, девочка настойчиво ищет еду, при этом аппетит избирательный, выдерживает до 4 часов без еды.
В возрасте 7 лет впервые обратились в ГНЦ ФГБУ «НМИЦ эндокринологии им. акад. И.И. Дедова» с жалобами на быстропрогрессирующую прибавку веса. При осмотре: рост — 122,5 см, SDS роста +0,56, вес — 46 кг, SDS ИМТ +4,04, Таннер 1 (В1, Р1), Ме abs.
Обращали на себя внимание следующие фенотипические особенности: широкий, часто открытый рот, беспричинный смех, аффективное поведение (счастливый внешний вид), плосковальгусные стопы, ходьба со специфической установкой рук, гиперсаливация (рис. 3).
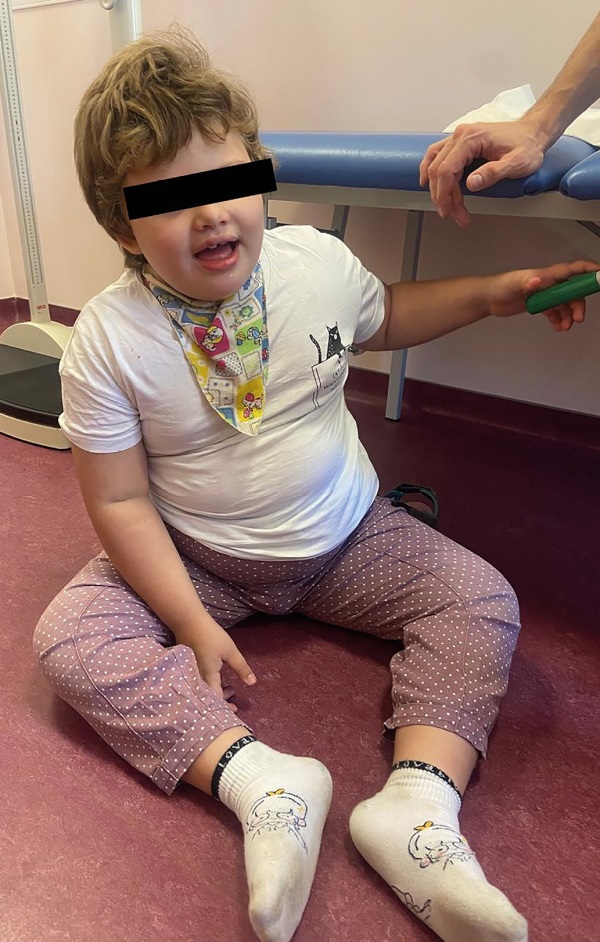
Рисунок 3. Фенотипические особенности у девочки с синдромом Ангельмана: широкий, часто открытый рот, счастливый внешний вид, плосковальгусные стопы, морбидное ожирение.
С учетом жалоб, анамнеза, фенотипических особенностей, был заподозрен синдром Ангельмана, направлена на генетическое исследование.
По результатам МЧ-MLPA, выполненной на базе лаборатории эпигенетики ожирения и диабета ФГБНУ «МГНЦ имени академика Н.П. Бочкова», у пациентки выявлено гипометилирование SNRPN, характерное для синдрома Ангельмана (рис. 4). Гипометилирование, по результатам МЧ-MLPA, может быть последствием как однородительской дисомии (ОРД), так и изолированного изменения метилирования (эпимутации). Для проведения дифференциальной диагностики молекулярной причины гипометилирования следует проводить поиск однородительской дисомии. По результатам микросателлитного анализа, выполненного на базе лаборатории молекулярно-генетической диагностики 2 ФГБНУ «МГНЦ им. академика Н.П. Бочкова», определена однородительская изодисомия (ОРД) отцовского происхождения, характерная для синдрома Ангельмана.
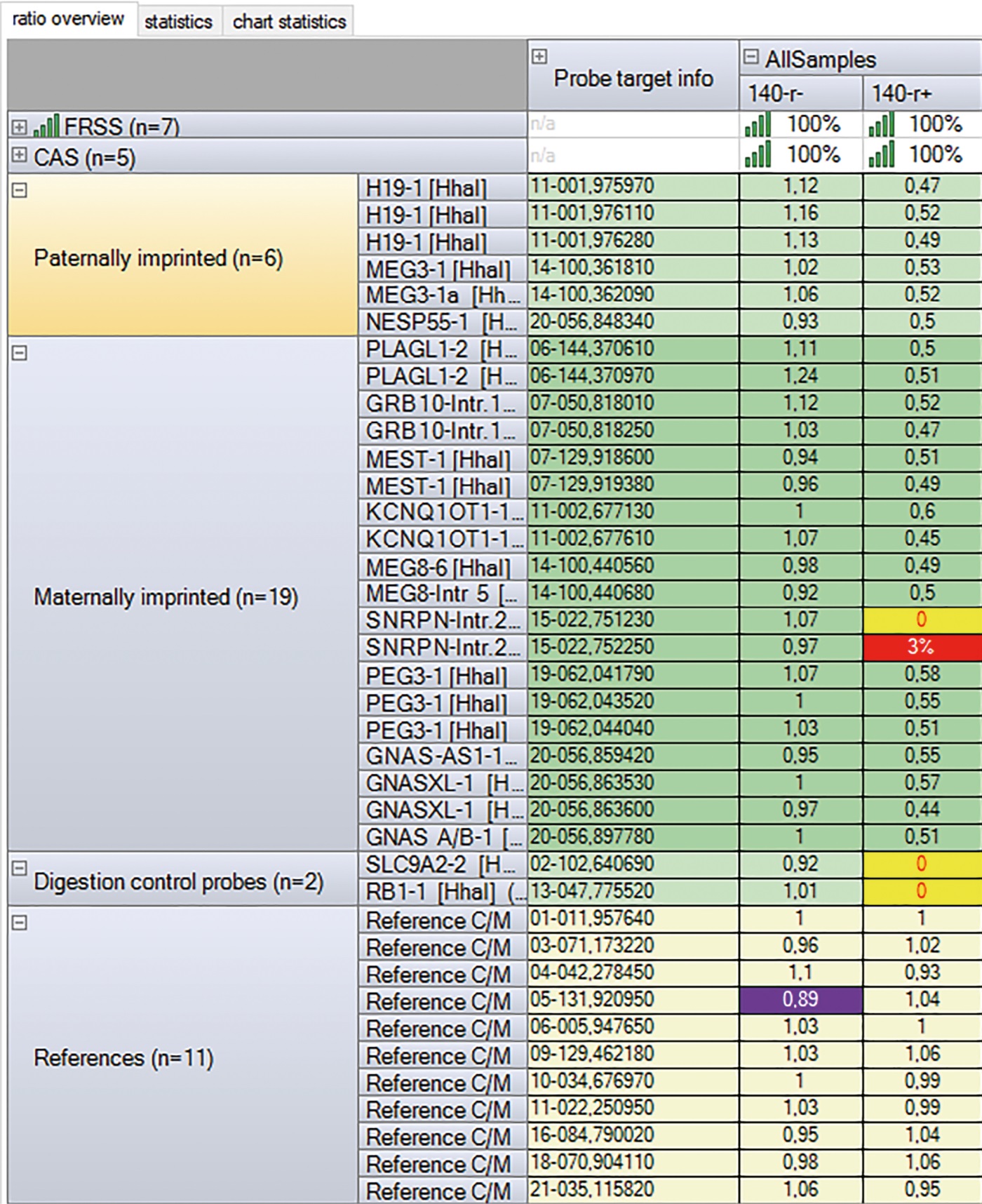
Рисунок 4. Результаты МЧ-MLPA пациентки С (№140). Столбец «140—r-» — результат анализа копийности; референсный интервал: 0,80–1,20. Столбец «140—r+» — результат анализа метилирования; референсный интервал: 0,40–0,65. Выявлено гипометилирование SNRPN (2 зонда из 2).
По результатам дообследования, из осложнений ожирения у девочки диагностированы инсулинорезистентность по данным ОГТТ (индекс Matsuda 2.02, максимальное значение уровня инсулина — 387,8 мкЕ/мл), неалкогольная жировая болезнь печени в стадии жирового гепатоза. Данные биохимических и гормональных исследований представлены в таблице 1. С метаболической целью рекомендована терапия препаратом метформина гидрохлорида в дозе 1000 мг/сут на фоне рационализации питания и адекватной физической нагрузки.
Таблица 1. Результаты обследований
|
Показатель |
Значение у пациента |
Норма |
|
Холестерин общий, ммоль/л |
3,58 |
3,3–5,2 |
|
Триглицериды, ммоль/л |
1,06 |
0,1–1,7 |
|
Холестерин ЛПНП, ммоль/л |
2,05 |
1,1–3 |
|
Холестерин ЛПВП, ммоль/л |
1,054 |
0,9–2,6 |
|
АСТ, Ед/л |
20,4 |
15–60 |
|
АЛТ, Ед/л |
20,2 |
7–35 |
|
Железо, мкмоль/л |
17,9 |
9–30,4 |
|
Ферритин, нг/мл |
54,6 |
20–200 |
|
Гликированный гемоглобин, % |
5,1 |
4–6 |
|
ТТГ, мМЕ/л |
2,16 |
0,51–4,82 |
|
свТ4, пмоль/л |
11,75 |
11,2–18,6 |
|
Кортизол, (кровь), утро, нмоль/л |
473,6 |
77–630 |
|
ИПФР-1, нг/мл |
231 |
24–396 |
Мальчик, 3 года. Родился от 1-й беременности, протекавшей на фоне отслойки плаценты на 10-й неделе гестации. Роды на 38-й неделе, самостоятельные. При рождении: вес — 3740 г (SDS 1,17), длина тела — 54 см (SDS 2,33). Ранний постнатальный период без особенностей. Наследственный анамнез: по ожирению и эндокринопатиям не отягощен. При рождении отмечался левосторонний крипторхизм (в 6 месяцев проведена орхипексия). Также обращала на себя внимание мышечная гипотония.
В возрасте 1 года 4 месяцев в связи с появлением нарушения коммуникации со сверстниками и взрослыми был консультирован неврологом, проведена диагностика по методике АДОС-2, в результате которой выявлена вероятность аутистических расстройств от умеренной до высокой степени.
В дальнейшем мальчику был установлен диагноз «Аутизм. Синдром мышечного гипотонуса», рекомендована АВА-терапия.
Мальчик был направлен на консультацию к генетику.
По результатам цитогенетического исследования — кариотип 46, ХY нормальный мужской.
Пациенту было проведено глубокое секвенирование ряда генов, включенных в большую неврологическую панель (лаборатория ООО «Геномед»), в результате чего был выявлен гетерозиготный нонсенс вариант NM_019066.5:c.2457T>A (p.Tyr819*) в гене MAGEL2. Патогенные варианты в этом гене описаны у пациентов с синдромом Шаафа-Янг. Вариант не зарегистрирован в популяционной базе данных gnomAD v2.1.1. На основании критериев PVS1 и PM2 (ACMG guideline 2015) вариант был расценен как вероятно патогенный. Сегрегационный анализ выявил носительство данного варианта у здорового отца. Кроме того, дальнейший анализ сегрегации показал, что бабушка пробанда по отцовской линии также является здоровым гетерозиготным носителем выявленного варианта. Поскольку ген MAGEL2 в норме экспрессируется с отцовского аллеля, для подтверждения патогенности варианта c.2457T>A был проведен РНК-анализ, позволяющий оценить, какой из аллелей гена MAGEL2 (мутантный или аллель дикого типа) экспрессируется у пробанда и у его отца.
Для этого в лаборатории функциональной геномики ФГБНУ «МГНЦ имени академика Н.П. Бочкова» был выполнен ОТ-ПЦР тотальной РНК, полученной из фибробластов кожи пробанда и его отца с последующим глубоким секвенированием продуктов амплификации.
Проведенный анализ последовательности мРНК гена MAGEL2 показал, что у пробанда экспрессируется один аллель, содержащий вариант c.2457T>A, в то же время у отца пробанда была обнаружена экспрессия аллеля дикого типа (рис. 5).
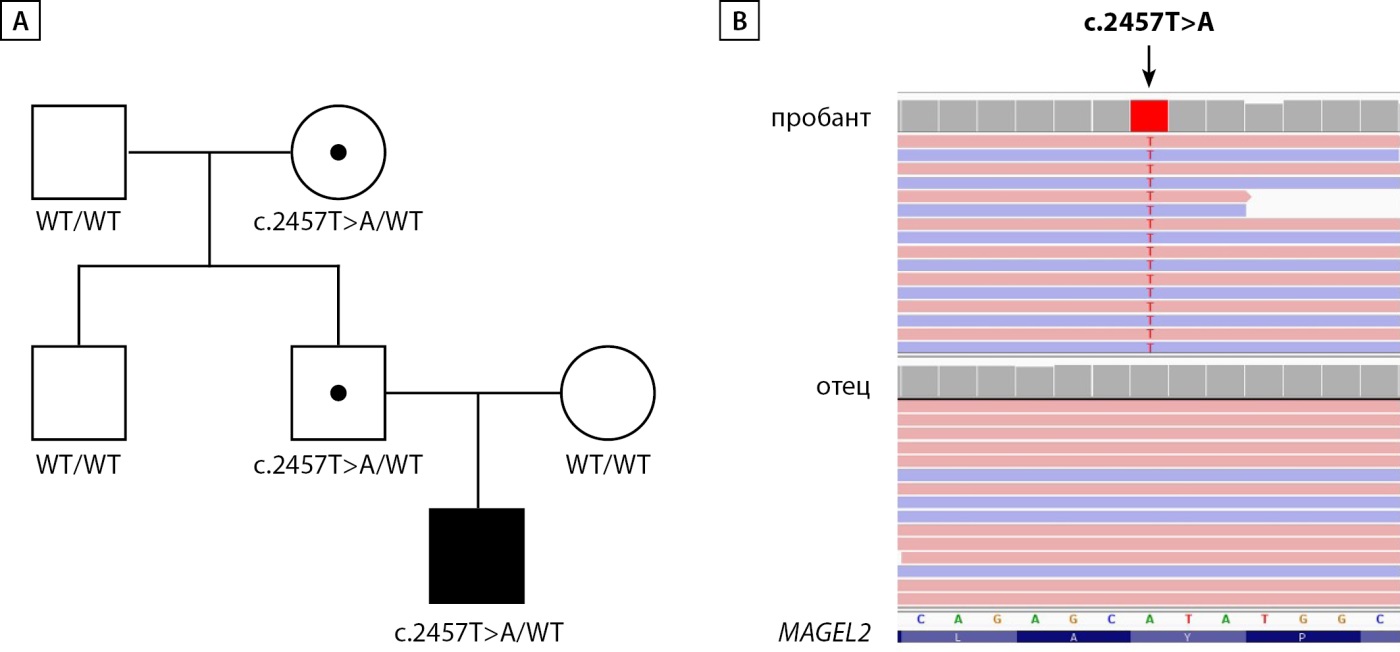
Рисунок 5. Молекулярно-генетический анализ пациента с нонсенс-вариантом c.2457T>A в гене MAGEL2.
A. Наследование варианта c.2457T>A в семье пробанда в трех поколениях. B. Визуализация данных глубокого секвенирования кДНК гена MAGEL2 c помощью IGV браузера иллюстрирует у пробанда экспрессию аллеля, содержащего вариант c.2457T>A, а у отца — экспрессию аллеля дикого типа по данному варианту.
СШЯ — аутосомно-доминантное заболевание, при котором мутация может наследоваться от фенотипически здорового отца, который, в свою очередь, унаследовал ее от фенотипически здоровой матери, поскольку ген MAGEL2 импринтирован по материнской линии и экспрессируется с аллеля отцовского происхождения. В отличие от классических аутосомно-доминантных заболеваний, фенотип СШЯ может пропускать несколько поколений, пока мутация находится на материнской (импринтированной) хромосоме, что обуславливает наличие фенотипических признаков у пробанда и отсутствие таковых у его отца и бабушки по отцовской линии.
Впервые обследован в ГНЦ ФГБУ «НМИЦ эндокринологии им. академика И.И. Дедова» в возрасте 3 лет с жалобами на особенности поведения и снижение чувства насыщения, эпизоды кетотических гипогликемий.
При осмотре: рост — 91,5 см, SDS роста -0,81, вес — 14 кг, SDS ИМТ +0,79. Половые органы сформированы правильно, по мужскому типу, Таннер 1 (G1, P1), яички в мошонке, volume testes D=S= 1.5 ml. Фенотипические особенности: форма головы долихоцефальная, форма лица квадратная, слегка нависающий высокий лоб, пухлые щеки, тонкая верхняя губа, горизонтальная складка на подбородке, диспластичные ушные раковины, конусовидные пальцы рук.
В гормональном профиле (венозная плазма натощак) отмечается эутиреоз, референсный уровень ИФР-1, АКТГ, кортизола и паратгормона, адекватно сниженный уровень инсулина на фоне гипогликемии (табл. 2).
Таблица 2. Результаты обследований
|
Показатель |
Значение у пациента |
Норма |
|
Глюкоза (сыворотка), ммоль/л |
2,33 |
3,1–6,1 |
|
Холестерин общий, ммоль/л |
4,74 |
3,3–5,2 |
|
Триглицериды, ммоль/л |
0,56 |
0,1–1,7 |
|
Холестерин ЛПНП, ммоль/л |
3 |
1,1–3 |
|
Холестерин ЛПВП, ммоль/л |
1,492 |
0,9–2,6 |
|
АСТ, Ед/л |
36,7 |
15–60 |
|
АЛТ, Ед/л |
11,3 |
7–35 |
|
Железо, мкмоль/л |
22,5 |
10,6–31,3 |
|
Ферритин, нг/мл |
37,7 |
20–200 |
|
Гликированный гемоглобин, % |
5,5 |
4–6 |
|
ТТГ, мМЕ/л |
2,414 |
0,64–5,76 |
|
свТ4, пмоль/л |
12,46 |
11,5–20,4 |
|
Кортизол, (кровь), утро, нмоль/л |
311 |
77–630 |
|
ИПФР-1, нг/мл |
43,96 |
11–233 |
|
Инсулин, мкЕ/мл |
1,31 |
2,6–24,9 |
|
Паратгормон, пг/мл |
17,51 |
15–65 |
|
АКТГ (утро), пг/мл |
9,38 |
7,2–63,3 |
При дальнейшем опросе мамы было выяснено, что мальчик находится на кетодиете ввиду избежания увеличения веса.
Выявленная гипогликемия была расценена как кетотическая, развившаяся на фоне специфической диеты. Проведена консультация диетолога, рекомендовано добавление в рацион сложноусвояемых углеводов. Учитывая возможность развития гипопитуитаризма и ожирения на фоне полифагии в рамках данного синдрома, рекомендовано дальнейшее динамическое наблюдение детским эндокринологом.
Риск повторного рождения ребенка с СШЯ в данной семье — 50% ввиду носительства мутации у отца. Если у ребенка получится сохранить репродуктивную функцию, то с 50%-ной вероятностью он будет передавать этот вариант своим детям и с 50%-ной вероятностью у них будет СШЯ.
Мальчик, 2 года 5 месяцев. Родился от 2-й беременности, протекавшей на фоне инфекции COVID-19 в 3-м триместре гестации. Роды на 39-й неделе, самостоятельные. При рождении: вес — 2146 г (SDS -3.19 SD), длина тела — 47 см (SDS -1.61 SD).
Наследственный анамнез: мама и папа — родственники 3-й степени родства. Мама — 164 см, 56 кг (ИМТ 20,8 — норма); папа — 178 см, 70 кг (ИМТ 22,1 — норма), родной брат (4 года) — задержка речевого развития, дефицит веса; у прабабушки по материнской линии сахарный диабет 2 типа.
При рождении был установлен диагноз «Задержка внутриутробного развития плода, гидроцефалия, синдром мышечной дистонии». Также отмечался двусторонний крипторхизм — в возрасте 6 месяцев был проведен курс терапии ХГЧ (хорионический гонадотропин человека) без положительной динамики, в связи с чем в 1,5 года проведена двусторонняя орхипексия.
Развитие на первом году жизни с задержкой: голову держит с 4-х месяцев, переворачивается на живот с 8 месяцев, переворачивается на спину с 10 месяцев, ползает по-пластунски с 1,5 года.
С 1-го года жизни наблюдается неврологом по поводу органического поражения головного мозга смешанного генеза (последствия перенесенной церебральной гипоксии, расширение межполушарной щели, умеренная вентрикуломегалия боковых желудочков), синдрома двигательных нарушений (диффузная мышечная гипотония, гипермобильность суставов), задержки психоречевого и моторного развития.
В возрасте 6 месяцев у мальчика был выявлен транзиторный субклинический гипотиреоз — отмечалось повышение уровня ТТГ до 15,4 мкМЕ/мл (норма 0,73–8,35) при референсном уровне свТ4 (12,7 пмоль/л при норме 11,9–25,6) и антител к тиреопероксидазе (менее 9 МЕ/мл при норме 0–34), инициирована терапия левотироксином натрия в дозе 12,5 мкг/сут, которую получал в течение 10 месяцев, после отмены терапии отмечался эутиреоз.
В возрасте 1 года 7 месяцев консультирован генетиком, учитывая наличие гипостратуры, грубой задержки моторного развития, выраженной диффузной мышечной гипотонии, гидроцефалии, двустороннего крипторхизма, фенотипических особенностей (редкие волосы на голове, высокий лоб, выступающие лобные бугры, миндалевидный разрез глаз, голубые склеры, треугольный подбородок, срединная ладонная борозда на обеих кистях, оттопыренные, чашеобразные и низкопосаженные ушные раковины, отсутствие верхней ножки противозавитка на обеих ушных раковинах, короткая шея, вальгусная деформация стоп, наложение второго пальца на третий на обеих стопах, неполная кожная синдактилия), было рекомендовано проведение цитогенетического и молекулярно-генетического исследования.
По результатам цитогенетического исследования — кариотип 46, ХY нормальный мужской.
По результатам МЧ-MLPA, выполненной на базе лаборатории эпигенетики ожирения и диабета ФГБНУ «МГНЦ имени академика Н.П. Бочкова», выявлены изменения, характерные для синдрома Темпл — определено гипометилирование MEG3 и гиперметилирование MEG8, выявлена делеция 14q32.2.
В возрасте 2 лет проведена консультация офтальмолога — установлен диагноз «Фоновая ретинопатия и ретинальные сосудистые изменения». При офтальмоскопии OU диск зрительного нерва розовый, границы четкие, макулярная зона без патологии, сосуды спазмированы, периферия без патологических изменений.
Мама ребенка неоднократно отмечала эпизоды состояний, клинически похожих на гипогликемию — сложность пробуждения, вялость, которые купировались приемом сладкого чая, однако ребенку в эти моменты глюкоза не измерялась.
Также у мальчика отмечается персистирующая железодефицитная анемия, по поводу которой периодически получает терапию препаратами трехвалентного железа со слабым положительным эффектом — уровень гемоглобина повышается, однако стабилизации показателей не происходит.
Впервые обследован в ГНЦ ФГБУ «НМИЦ эндокринологии им. академика И.И. Дедова» в возрасте 2 лет 5 месяцев с жалобами на дефицит веса, низкую прибавку массы тела, склонность к гипогликемиям.
При осмотре: рост — 78 см, SDS роста -3,13, вес — 6,7 кг, SDS ИМТ -7. Половые органы сформированы правильно, по мужскому типу, Таннер 1 (G1, P1), яички в мошонке, volume testes D=S= 1.0 ml. Обращала на себя внимание выраженная диффузная мышечная гипотония, задержка моторного развития — ребенок сидит, но не стоит и не ходит, плохо держит предметы в руках, отмечаются проблемы с жеванием, трудности артикуляции.
По результатам лабораторных исследований (табл. 3) сохраняется железодефицитная анемия средней степени тяжести, рекомендовано продолжит терапию препаратами железа. В гормональном исследовании (табл. 3) отмечается эутиреоз, референсный уровень ИФР-1, кортизола и пролактина.
Таблица 3. Результаты обследований
|
Показатель |
Значение у пациента |
Норма |
|
Эритроциты (кровь), 10^12 кл/л |
3,34 |
3,8–5,2 |
|
Гемоглобин, г/л |
89 |
115–150 |
|
Гематокрит, % |
25,8 |
35–46 |
|
Глюкоза (сыворотка), ммоль/л |
3,52 |
3,1–6,1 |
|
АСТ, Ед/л |
60 |
15–60 |
|
АЛТ, Ед/л |
15,12 |
7–35 |
|
Железо, мкмоль/л |
10,5 |
10,6–31,3 |
|
Ферритин, нг/мл |
18 |
20–200 |
|
Креатинин, мкмоль/л |
17,7 |
27–62 |
|
ТТГ, мкМЕ/л |
3,78 |
0,7–6,0 |
|
Т4 свободный, нг/дл |
1,1 |
0,9–1,7 |
|
Кортизол, (кровь), утро, мкг/дл |
12,4 |
1,73–10,76 |
|
ИПФР-1, нг/мл |
158,6 |
11–233 |
|
Инсулин, мкЕ/мл |
0,34 |
2,6–24,9 |
|
Пролактин, нг/мл |
11,11 |
3,9–20,1 |
Учитывая выраженную диффузную мышечную гипотонию, критический дефицит массы тела, задержку роста и моторного развития, для улучшения ростового прогноза, композиционного состава тела и мышечного тонуса была рекомендована терапия рекомбинантным гормоном роста в дозе 0,033 мг/кг/сут.
Риск повторного рождения в данной семье ребенка с СТ в случае делеции и ОРД зависит от обследования родителей, в случае эпимутации — низкий, но выше популяционного; также важно, что если у мальчика получится сохранить репродуктивную функцию, то он будет в 50% передавать хромосому 14 с делецией и это, с учетом определенного импринтированного статуса генов в этом кластере, чревато риском 50% рождения ребенка с СТ.
Анализ современных данных демонстрирует, что заболевания, обусловленные нарушением геномного импринтинга, играют важную роль в понимании патогенеза ожирения, особенно в детском возрасте. Такие синдромы, как Прадера–Вилли, Ангельмана, Шаафа-Янг, Темпл, псевдогипопаратиреоз 1а и 1b типов, иллюстрируют, как дисрегуляция экспрессии импринтированных генов может приводить к нарушению энергетического баланса, гиперфагии, снижению физической активности и аномальному распределению жировой ткани. Важной особенностью этих заболеваний является то, что ожирение возникает на фоне комплексных нейроэндокринных и метаболических нарушений, в основе которых лежат дефекты импринтинга. Ключевыми мишенями являются гены, регулирующие гипоталамическую функцию, ростовые факторы, сигнальные каскады инсулина и лептина.
При синдроме Ангельмана выделяют следующие причины развития заболевания: делеция региона 15q11.2–13 хромосомы 15 материнского происхождения — более 70% случаев; мутация гена UBE3A (локус 15q11.2), расположенного на хромосоме 15, унаследованной от матери — 20–25% случаев; однородительская дисомия (ОРД) региона 15q11.2–13 отцовского происхождения — 5–7% случаев; дефект центра импринтинга — около 3% случаев. Известно, что в хромосоме 15 только материнский аллель гена UBE3A функционально активен и только он экспрессируется в головном мозге. Отцовский аллель гена UBE3A подвергается функциональному выключению за счет процесса антисмысловой транскрипции [12]. Большие (5–7 Mб) делеции 15q11.2–13, включающие материнский аллель гена UBE3A, являются причиной недостаточной продукции специфичных для нейронов головного мозга белков Ube3a [13]. Эти белки (ферменты) необходимы для функционирования системы убиквитин–протеасома, регулирующей многие клеточные процессы, в частности обеспечивающей деградацию и устранение поврежденных белков [14]. Экспрессия UBE3A имеет решающее значение для синаптической пластичности, синаптогенеза, морфологической зрелости нейронов и поддержания надлежащего уровня нейромедиатора гамма-аминомасляной кислоты. Недостаточная экспрессия белка Ube3a также возникает при мутации материнского аллеля гена UBE3A, при однородительской (отцовской) дисомии 15q11.2–13 и при дефекте центра импринтинга.
Дети с СА рождаются с нормальными антропометрическими показателями. К ранним симптомам заболевания, заметным с первых недель/месяцев жизни, можно отнести проблемы с кормлением (несогласованные движения языка, мышц губ и щек, участвующих в сосании), страбизм, мышечную гипотонию, задержку двигательного развития. Нарушения психомоторного развития и особенности поведения (аффективное поведение, гиперактивность, улыбчивость, частый и легко провоцируемый смех) чаще возникают во втором полугодии жизни, а апраксия движений рук и стереотипии рук — ближе к году.
Кроме того, с раннего возраста может дебютировать рефрактерная эпилепсия с характерными патологическими изменениями на электроэнцефалограмме (ЭЭГ) [9–11]. Ранний дебют эпилепсии (на 1-м году жизни) отмечен только у отдельных пациентов (у 4, в 9% всех случаев с эпилепсией), хотя, по данным литературы, около 25% пациентов заболевают эпилепсией в возрасте до одного года. Патологические изменения на ЭЭГ, по данным литературы, могут быть зарегистрированы у большинства пациентов с синдромом Ангельмана, в том числе у детей, не имевших эпилептических приступов. Уже в возрасте 4–9 месяцев у детей с СА появляются характерные изменения ЭЭГ в виде высокоамплитудных вспышек медленных волн зазубренного/расщепленного характера (notched delta), которые чаще регистрируются в затылочных отделах, но могут быть и диффузными.
Также данные пациенты имеют ряд фенотипических проявлений: микроцефалия, уплощенный затылок, широкий рот, редкие зубы, выступающий язык, прогнатия. Однако эти особенности становятся заметными лишь к 2–3 годам.
В 2021 г. Горчхановой З.К. и соавт. был опубликован анализ клинических проявлений СА у детей [18]. В исследование было включено 60 детей, которым был установлен диагноз по международным клиническим критериям и результатам генетического исследования. В 100% случаев у пациентов отмечалась задержка психического и моторного развития, а также проблемы с речью. У 17% пациентов отмечались трудности с кормлением на первом году жизни, а в 53% — мышечная гипотония. Наиболее частыми неврологическими проявлениями были стереотипии рук (90%), тремор (56%), апраксия движений рук (93%), атаксия (83%) и эпилептические приступы (72%). Из особенностей поведения в 97% обращало на себя внимание аффективное поведение (счастливый внешний вид, дружелюбие), в 70% отмечался беспричинный смех, а в 58% — гиперактивность. В 65% случаев у наблюдавшихся пациентов уже на первом году жизни отмечалось нарушение сна (беспокойный сон, кратковременный сон или сон с частыми пробуждениями), что может быть обусловлено сниженным уровнем мелатонина в сыворотке крови у пациентов с СА [66].
В 2023 г. группа ученых из Университетского медицинского центра Роттердама провела исследование гиперфагии, роста и полового созревания у детей с СА [19]. Из 145 обследованных детей с СА у 23% отмечалась избыточная масса тела и у 20% — ожирение.
Ожирение при синдроме Ангельмана не является типичным симптомом, как, например, при синдроме Прадера-Вилли. Оно может быть связано с нейропсихологическими особенностями, нарушениями регуляции сна, приемом антиэпилептических препаратов или снижением двигательной активности. Согласно источникам, пациенты с СА ввиду ОРД имеют более высокий риск ожирения, чем с делеционным типом [52][53][54][55][56]. Такие пациенты чаще страдают гиперфагией, а их вес значительно увеличивается в основном после 2-летнего возраста, по сравнению с другими молекулярными причинами СА [57]. У некоторых пациентов увеличение массы тела может быть очевидным уже на первом году жизни, что доказывает целесообразность рассмотрения диагноза СА у детей раннего возраста с ожирением и задержкой развития, особенно в тяжелой форме [58].
Несмотря на редкость гиперфагии при данном синдроме, важно внимательно следить за пищевыми привычками пациентов, чтобы предотвратить избыточное потребление пищи и связанные с этим риски ожирения и метаболических нарушений. Комплексный подход, включающий диетическое сопровождение и мониторинг общего состояния пациента, играет ключевую роль в профилактике гиперфагии и поддержании оптимального здоровья при синдроме Ангельмана.
Для постановки диагноза детям с подозрением на СА проводится молекулярно-генетическое исследование. Диагностический поиск начинают с анализа метилирования ДНК хромосомы 15, в области 15q11.2–13. Дифференциальное метилирование указанного района обеспечивает основу для диагностического тестирования на СА. В норме у здоровых людей метилирован материнский аллель и не метилирован отцовский аллель SNRPN в регионе 15q11.2–13, что можно определить с помощью исследования промоторной области гена SNRPN путем МЧ-MLPA. При синдроме Ангельмана выявляется гипометилирование SNRPN, так как в результате делеции, ОРД или эпимутации метилированный статус материнского аллеля SNRPN отсутствует. Нормальный профиль метилирования не исключает СА, потому что в 11% случаев этот синдром обусловлен мутацией гена UBE3A и не сопровождается изменением метилирования [50]. Раннюю постановку диагноза СА затрудняет сходство его клинических проявлений с другими заболеваниями нервной системы, в том числе обусловленными наследственными нарушениями обмена веществ и различными хромосомными аномалиями. В ряде случаев дети с синдромом Ангельмана длительно наблюдаются неврологами с диагнозами детского церебрального паралича, атонически-астатической формы и эпилепсии. В то же время знание клинических особенностей синдрома Ангельмана дает возможность более раннего установления диагноза.
Своевременная постановка диагноза важна не только для правильной тактики оказания помощи ребенку, но и для оценки репродуктивных рисков в семье. СА по причине ОРД предполагает обязательное генетическое консультирование с применением кариотипирования родителей пробанда: в случае выявления носительства отцом робертсоновской транслокации rob (15;15) риск повторного рождения ребенка с СА близок к 100%. СА по причине делеции требует цитогенетического обследования матери пробанда; СА по причине унаследованной мутации UBE3A или делеции центра импринтинга создает риск повтора рождения ребенка с СА в 50%. В случае нормального результата цитогенетического исследования родителей риск повторного рождения ребенка с СА составляет <1% [50].
Медикаментозное лечение при СА включает контроль эпилептических приступов, коррекцию поведенческих нарушений и расстройств сна. Лечение эпилепсии — основная задача терапии при СА. Однако уникальная эпигенетика гена UBE3A предлагает потенциальный терапевтический путь. Ученые Вихма Х., Ли К., Велтон-Арндт и др. предположили, что реактивация отцовской копии гена UBE3A с помощью малой молекулы (S)-PHA533533 может лежать в основе лечения синдрома Ангельмана. Отцовский аллель гена UBE3A эпигенетически подавляется антисмысловой транскрипцией в нейронах, а мутация материнского аллеля UBE3A является одной из причин СА. Эти процессы ведут к устранению избытка белка Ube3a в головном мозге. Реактивация отцовского аллеля гена UBE3A может быть перспективным направлением в лечении СА [20]. Таким образом, в настоящее время ранняя диагностика становится актуальной задачей в связи с интенсивными изысканиями в области генной терапии заболевания.
Синдром Шаафа-Янг вызван патогенными вариантами в гене MAGEL2, находящемся в кластере импринтированных генов в районе 15q11.2-13 [21]. MAGEL2 импринтирован по материнской линии и, таким образом, экспрессируется с отцовского аллеля. Ген MAGEL2 выполняет плейотропные функции в организме человека, особенно в гипоталамусе. Предполагается, что MAGEL2 является специфическим для млекопитающих регулятором нейроэндокринной функции гипоталамуса, настраивая гипоталамическую регуляцию физиологического гомеостаза и поведения для адаптации к сигналам окружающей среды [41]. Также MAGEL2 играет ключевую роль в нейрогенезе [42].
Клинически СШЯ сильно варьирует не только в зависимости от каждого конкретного случая, но и в течение всей жизни пациентов. Внутриутробно СШЯ может проявляться многоводием и снижением двигательной активности плода, однако при рутинном пренатальном обследовании заболевание выявляется крайне редко [23–29]. Для постнатального периода характерны диффузная мышечная гипотония, слабый сосательный рефлекс, респираторный дистресс-синдром с эпизодами апноэ сна [22][29]. Одним из ранних диагностически значимых признаков является наличие контрактур суставов, а в более тяжелых случаях — множественного врожденного артрогрипоза, развивающегося в результате снижения или отсутствия двигательной активности плода [30–32]. По мере взросления становятся очевидными психоневрологические особенности пациентов: задержка интеллектуального развития разной степени выраженности, РАС, а также импульсивность, обсессивно-компульсивное поведение, склонность к манипуляциям, аутоагрессия и повышенная тревожность [22][33]. У большинства пациентов с СШЯ наблюдается грубая гипоталамическая дисфункция, которая проявляется в виде дефицита гормона роста, гипогонадотропного гипогонадизма, нестабильности температуры тела и нарушения циркадных ритмов [22][24][34]. Внешне у пациентов с СШЯ нередко отмечаются неспецифические краниофациальные особенности, такие как квадратная форма головы и лица, гипертелоризм глаз, антимонголоидный разрез глаз, низко посаженные уши, выступающая переносица, короткий фильтр, макроцефалия и микрогнатия [35–37]. Полифагия и ожирение встречается у меньшинства молодых пациентов с CШЯ и, как правило, появляются в более старшем возрасте, в отличие от пациентов с СПВ [25][38][39].
На сегодняшний день специфической терапии для СШЯ не существует. Терапия, предоставляемая пациентам, включает логопедию, психотерапию и психиатрическое лечение, физиотерапию, кормление через желудочный или назогастральный зонд, а также заместительную гормональную терапию.
Alicia F Juriaans, Gerthe F Kerkhof, et al. в своем исследовании, включившем 7 пациентов с СШЯ, которые получали терапию рекомбинантным гормоном роста на протяжении четырех лет, показали, что данная терапия приводит к значительному улучшению показателей SDS роста и ИМТ [43].
Продолжается активный поиск эффективной терапии для СШЯ. Окситоцин привлек значительное внимание исследователей как потенциальное терапевтическое средство для лечения РАС при СШЯ и СПВ, при этом клинические испытания дали неоднозначные результаты [44][45]. Генная терапия с использованием аденоассоциированного вируса Bdnf улучшала состав тела, расход энергии, метаболизм глюкозы и поведение у мышей с Magel2-null геном [46]. На мышиных моделях с Magel2-null геном был показан положительный эффект терапии сетмеланотидом (агонистом рецептора MC4) в виде уменьшения аппетита и увеличения расхода энергии, однако перенос этого в клиническую практику имеет особые сложности [47]. Также в виде перспективной терапевтической стратегии может рассматриваться антисенс-терапия [48].
Важное значение имеет генетическое консультирование семей с ребенком, имеющим СШЯ. Если у отца обнаружена аналогичная мутация, то риск рождения ребенка с СШЯ в этой семье равен 50%.
Синдром Темпл может быть вызван материнской ОРД по хромосоме 14 (72–78%); изолированной потерей метилирования в районе MEG3-DMR (12–20%); делецией 14q32.2 отцовского происхождения (10%), которые приводят к увеличению экспрессируемых материнских генов и подавлению экспрессируемых отцовских генов в импринтированном домене 14q32.2 [59].
Alicia F Juriaans, Gerthe F Kerkhof и соавт. в 2022 г. описали 15 пациентов с СТ [63]. У восьми пациентов была ОРД(14)мат, а у семи — дефект метилирования. Наиболее распространенными симптомами были задержка внутриутробного развития (100%), мышечная гипотония (100%), преждевременное половое развитие (89%), низкий вес при рождении (67%), проблемы с кормлением (53%) и психо-поведенческие особенности (53%). У семи пациентов (47%) отмечалась гиперфагия, у пяти (33%) — избыточный вес или ожирение, медиана SDS процента жировой массы (ЖМ%) составила 2,53 [ 2,26; 2,90], а медиана SDS тощей массы тела (ТМТ) -2,03 [ -3,22; -1,28]. Не было никаких существенных различий в клинических характеристиках между пациентами с ОРД(14)мат и дефектом метилирования.
А в 2024 г. Tomoe Ogawa и соавт. опубликовали исследование, описывающее 60 пациентов из Японии [64]. Мышечная гипотония, постнатальный низкий рост и гонадотропинзависимое преждевременное половое развитие были выявлены у 61,5%, 87,0% и 86,0% пациентов соответственно. Кроме того, исследование показало, что 20,0% пациентов имели ожирение, у 26,5% пациентов в возрасте старше 6 лет была гиперхолестеринемия, у 12,8% пациентов в возрасте старше 9 лет — сахарный диабет.
Основные терапевтические действия при СТ направлены на коррекцию преждевременного полового развития (терапия пролонгированными аналогами гонадотропин-рилизинг гормона), низкорослости (терапия рекомбинантным гормоном роста) и проблем нейроразвития [64]. Для лечения ожирения при СТ специфической терапии в настоящее время не существует, рекомендуется диетотерапия и лечебная физкультура. При наличии метаболических осложнений, таких как дислипидемия и сахарный диабет, может рассматриваться терапия ингибиторами гидроксиметилглутарил-коэнзим А (ГМГ-КоА)-редуктазы, ингибиторами натрий-глюкозного котранспортера 2-го типа и инсулинотерапия [64]. Важно, что ведение данной группы пациентов должно осуществляться многопрофильной командой, чтобы охватить все важные проблемы.
Особый интерес представляет возможная связь между частичными нарушениями импринтинга и фенотипами ожирения у лиц без ярко выраженных синдромов. Это открывает перспективу для пересмотра патогенеза ожирения как заболевания, в определенных случаях обусловленного не только диетой и образом жизни, но и тонкими нарушениями эпигенетического регулирования.
Несмотря на то, что данные синдромы редки, их изучение позволяет лучше понять фундаментальные эпигенетические механизмы, управляющие энергетическим гомеостазом. Кроме того, наблюдаются данные о возможной роли мягких форм нарушений импринтинга в развитии ожирения у лиц без четко выраженного синдромального фенотипа. Это поднимает вопрос о том, могут ли эпигенетические механизмы импринтинга быть задействованы в патогенезе более распространенных форм ожирения, особенно с ранним началом и резистентностью к терапии.
Геномный импринтинг представляет собой важный, но часто недооцененный механизм, способствующий развитию ожирения. Клиническая диагностика и лабораторное подтверждение подобных нарушений имеют значение не только для понимания редких генетических синдромов, но и для поиска новых биомаркеров и терапевтических мишеней при более распространенных формах ожирения. Будущие исследования, направленные на картирование импринтинговых дефектов и оценку их влияния на метаболизм, могут значительно расширить существующие подходы к лечению и профилактике ожирения. Таким образом, болезни геномного импринтинга предоставляют уникальное окно в изучение эпигенетических основ ожирения, подчеркивая важность междисциплинарного подхода, объединяющего генетику, эндокринологию, педиатрию и молекулярную биологию.
Конфликт интересов. Авторы декларируют отсутствие конфликта интересов.
Участие авторов. Атавина-Ермакова Е.П., Васюкова О.В., Андреева М.А., Копытина Д.А. — клиническое обследование пациентов, поисково-аналитическая работа и подготовка финальной версии статьи; Панченко Е.Г. — проведение части клинического обследования, проведение генетического исследования, интерпретация результатов, поиск литературы, редактирование текста; Cимонова О.А. — проведение генетического исследования, интерпретация результатов; Удалова В.Ю. — интерпретация результатов, редактирование текста; Канивец И.В. — проведение части клинического обследования, поиск литературы, редактирование текста; Акимова Д.Б. — проведение генетического исследования, интерпретация результатов, редактирование текста; Скоблов М.Ю. — дизайн функционального исследования, поиск литературы, редактирование текста; Нагаева Е.В., Мокрышева Н.Г. — идея и дизайн исследования, редактирование текста, финальная редакция статьи.
Все авторы одобрили финальную версию статьи перед публикацией, выразили согласие нести ответственность за все аспекты работы, подразумевающую надлежащее изучение и решение вопросов, связанных с точностью или добросовестностью любой части работы.
Согласие пациентов. Законные представители пациентов подписали информированное согласие на публикацию персональной медицинской информации в журнале в обезличенной форме.
1. Miho Ishida, Gudrun E Moore. The role of imprinted genes in humans. Mol Aspects Med. 2013 Jul-Aug;34(4):826-40. doi: 10.1016/j.mam.2012.06.009. Epub 2012 Jul 4.
2. Назаренко С.А. Геномный импринтинг и его роль в этиологии наследственных болезней человека. Бюллетень сибирской медицины. 2004;3(3):8-17. https://doi.org/10.20538/1682-0363-2004-3-8-17
3. J. McGrath, D. Solter. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell, 37 (1984), pp. 179-183.
4. M.A. Surani, S.C. Barton, M.L. Norris. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature, 308 (1984), pp. 548-550.
5. D.P. Barlow, R. Stoger, B.G. Herrmann, K. Saito, N. Schweifer. The mouse insulin-like growth factor type-2 receptor is imprinted and closely linked to the Tme locus. Nature, 349 (1991), pp. 84-87
6. A. Lewis, W. Reik. How imprinting centres work. Cytogenet. Genome Res., 113 (2006), pp. 81-89
7. Debopam S. Epilepsy in Angelman syndrome: A scoping review. Brain Dev 2021; 43(1): 32–44. DOI: 10.1016/j.braindev.2020.08.014
8. Angelman H. ‘Puppet’ children. A Report on Three Cases. Development Med Child Neurol 2008. DOI: 10.1111/j.1469-8749.1965.tb07844.x
9. Pelc K., Boyd S.G., Cheron G., Dan B. Epilepsy in Angelman syndrome. Seizure 2008; 17:211–217. DOI: 10.1016/j.seizure.2007.08.004
10. Park S.H., Yoon J.R., Kim H.D., Lee J.S., Lee Y.M., Kang H.C. Epilepsy in Korean patients with Angelman syndrome. Korean J Pediatr 2012; 55: 171–176. DOI: 10.3345/kjp.2012.55.5.171
11. Thibert R.L., Larson M., Hsieh D.T., Raby A.R., Thiele E.A. Neurologic manifestations of Angelman syndrome. Pediatr Neurol 2013; 48(4): 271–279. DOI: 10.1016/j.pediatrneurol.2012.09.015
12. Meng L., Person R.E., Beaudet A.L. Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum Mol Genet 2012; 21: 3001–3012. DOI: 10.1093/hmg/dds130
13. Khatri N., Man H.Y. The Autism and Angelman Syndrome Protein Ube3A/E6AP: The Gene, E3 Ligase Ubiquitination Targets and Neurobiological Functions. Front Mol Neurosci 2019; 12:109. DOI: 10.3389/fnmol.2019.00109
14. Baloghova N., Lidak T., Cermak L. Ubiquitin Ligases Involved in the Regulation of Wnt, TGF-β, and Notch Signaling Pathways and Their Roles in Mouse Development and Homeostasis. Genes (Basel) 2019; 10(10): 815. DOI: 10.3390/genes10100815
15. Ramsden S.C., Clayton-Smith J., Birch R., Buiting K. Practice guidelines for the molecular analysis of Prader-Willi and Angelman syndromes. BMC Med Genet 2010; 11: 70. DOI: 10.1186/1471-2350-11-70
16. Duca D.G., Craiu D., Boer M., Chirieac S.M., Arghir A., Tutulan-Cunita A. et al. Diagnostic approach of Angelman syndrome. Maedica (Buchar) 2013; 8(4): 321–327
17. Bird L.M. Angelman syndrome: review of clinical and molecular aspects. Appl Cli
18. Горчханова З.К., Николаева Е.А., Боченков С.В., Белоусова Е.Д. Анализ клинических проявлений синдрома Ангельмана у детей. Российский вестник перинатологии и педиатрии. 2021;66(6):63-70. https://doi.org/10.21508/1027-4065-2021-66-6-63-70
19. Bindels-de Heus, K.G.C.B.; Hagenaar, D.A.; Dekker, I.; van der Kaay, D.C.M.; Kerkhof, G.F.; ENCORE Expertise Center for AS; Elgersma, Y.; de Wit, M.-C.Y.; Mous, S.E.; Moll, H.A. Hyperphagia, Growth, and Puberty in Children with Angelman Syndrome. J. Clin. Med. 2023, 12, 5981. https://doi.org/10.3390/jcm12185981
20. Vihma, H., Li, K., Welton-Arndt, A. et al. Ube3a unsilencer for the potential treatment of Angelman syndrome. Nat Commun 15, 5558 (2024). https://doi.org/10.1038/s41467-024-49788-8
21. Tim Schubert, Christian P Schaaf. MAGEL2 (patho‐)physiology and Schaaf–Yang syndrome. Dev Med Child Neurol. 2024 Jul 1;67(1):35–48. doi: 10.1111/dmcn.16018
22. Schaaf CP, Marbach F. Schaaf‐Yang Syndrome. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews®. Seattle (WA): University of Washington, Seattle, 2021.
23. Nunes S, Xavier M, Lourenço C, Melo M, Godinho C. Schaaf‐Yang syndrome: A real challenge for prenatal diagnosis. Cureus 2021; 13: e20414.
24. Marbach F, Elgizouli M, Rech M, et al. The adult phenotype of Schaaf‐Yang syndrome. Orphanet J Rare Dis 2020; 15: 294.
25. Fountain MD, Aten E, Cho MT, et al. The phenotypic spectrum of Schaaf‐Yang syndrome: 18 new affected individuals from 14 families. Genet Med 2017; 19: 45–52.
26. Mari A, Sartorio MUA, Degrassi I, et al. Late‐onset pyloric stenosis and intussusception with final diagnosis of food proteins' hypersensitivity in Schaaf‐Yang syndrome: A case report. JPGN Rep 2022; 3: e202.
27. Soden SE, Saunders CJ, Willig LK, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med 2014; 6: 265ra168.
28. de Andrade G, de Oliveira Silva T, do Nascimento I, Boath A, da Costa Cunha K, Chermont AG. Schaaf‐Yang syndrome: A novel variant in MAGEL2 gene in the first Brazilian preterm neonate. Int J Case Rep Images 2020; 11: 01144Z01GA202.
29. Xu N, Shi W, Cao X, et al. Preimplantation genetic testing (PGT) and prenatal diagnosis of Schaaf‐Yang Syndrome: A report of three families and a research on genotype‐phenotype correlations. J Clin Med Res 2023; 12: 1688.
30. McCarthy J, Lupo PJ, Kovar E, et al. Schaaf‐Yang syndrome overview: Report of 78 individuals. Am J Med Genet A 2018; 176: 2564–74.
31. Juriaans AF, Kerkhof GF, Hokken‐Koelega ACS. The spectrum of the Prader‐Willi‐like pheno‐ and genotype: A review of the literature. Endocr Rev 2022; 43: 1–18.
32. Mejlachowicz D, Nolent F, Maluenda J, et al. Truncating Mutations of MAGEL2, a Gene within the Prader‐Willi Locus, Are Responsible for Severe Arthrogryposis. Am J Hum Genet 2015; 97: 616–20.
33. Dötsch L, Matesevac L, Strong TV, Schaaf CP. Caregiver‐based perception of disease burden in Schaaf‐Yang syndrome. Mol Genet Genomic Med 2023; 11: e2262.
34. Powell WT, Schaaf CP, Rech ME, Wrede J. Polysomnographic characteristics and sleep‐disordered breathing in Schaaf‐Yang syndrome. Pediatr Pulmonol 2020; 55: 3162–7.
35. Patak J, Gilfert J, Byler M, et al. MAGEL2‐related disorders: A study and case series. Clin Genet 2019; 96: 493–505.
36. Alavanda C, Arslan Ateş E, Yavaş Abalı Z, Geçkinli BB, Turan S, Arman A. Two new cases with novel pathogenic variants reflecting the clinical diversity of Schaaf‐Yang syndrome. Clin Genet 2023; 104: 127–32.
37. Gregory LC, Shah P, Sanner JRF, et al. Mutations in MAGEL2 and L1CAM are associated with congenital hypopituitarism and arthrogryposis. J Clin Endocrinol Metab 2019; 104: 5737–50.
38. McCarthy JM, McCann‐Crosby BM, Rech ME, et al. Hormonal, metabolic and skeletal phenotype of Schaaf‐Yang syndrome: a comparison to Prader‐Willi syndrome. J Med Genet 2018; 55: 307–15.
39. Fountain MD, Schaaf CP. Prader‐Willi Syndrome and Schaaf‐Yang Syndrome: Neurodevelopmental Diseases Intersecting at the MAGEL2 Gene. Diseases 2016; 4. 10.3390/diseases4010002.
40. Дадали Е.Л., Маркова Т.В., Бостанова Ф.М. и др. Особенности клинических проявлений и генетических характеристик синдрома Шаафа–Янга у российских пациентов. Нервно-мышечные болезни 2024;14(1): 42–50. DOI: https://doi.org/10.17650/2222-8721-2024-14-1-42-50
41. Hoyos Sanchez MC, Bayat T, Gee RRF, Fon Tacer K. Hormonal imbalances in Prader‐Willi and Schaaf‐Yang syndromes imply the evolution of specific regulation of hypothalamic neuroendocrine function in mammals. Int J Mol Sci 2023; 24: 13109.
42. Lee S, Kozlov S, Hernandez L, et al. Expression and imprinting of MAGEL2 suggest a role in Prader‐willi syndrome and the homologous murine imprinting phenotype. Hum Mol Genet 2000; 9: 1813–9.
43. Juriaans AF, Kerkhof GF, Garrelfs M, Trueba‐Timmermans D, Hokken‐Koelega ACS. Schaaf‐Yang syndrome: Clinical phenotype and effects of 4 years of growth hormone treatment. Horm Res Paediatr 2023; 97: 148–56.
44. Althammer F, Muscatelli F, Grinevich V, Schaaf CP. Oxytocin‐based therapies for treatment of Prader‐Willi and Schaaf‐Yang syndromes: evidence, disappointments, and future research strategies. Transl Psychiatry 2022; 12: 318.
45. Ooi YP, Weng S‐J, Kossowsky J, Gerger H, Sung M. Oxytocin and Autism Spectrum Disorders: A Systematic Review and Meta‐Analysis of Randomized Controlled Trials. Pharmacopsychiatry 2017; 50: 5–13.
46. Queen NJ, Zou X, Anderson JM, et al. Hypothalamic AAV‐BDNF gene therapy improves metabolic function and ys in the Magel2‐null mouse model of Prader‐Willi syndrome. Mol Ther Methods Clin Dev 2022; 27: 131–48.
47. Bischof JM, Van Der Ploeg LHT, Colmers WF, Wevrick R. Magel2‐null mice are hyper‐responsive to setmelanotide, a melanocortin 4 receptor agonist. Br J Pharmacol 2016; 173: 2614–21.
48. Dhuri K, Bechtold C, Quijano E, et al. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J Clin Med Res 2020; 9. 10.3390/jcm9062004.
49. Schaaf C.P., Gonzalez-Garay M.L., Xia F. et al. Truncating mutations of MAGEL2 cause Prader–Willi phenotypes and autism. Nat Genet 2013;45 (11):1405–8. DOI: 10.1038/ng.2776
50. Dagli AI, Mathews J, Williams CA. Angelman Syndrome. 1998 Sep 15 [updated 2025 May 1]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1144/
51. Williams C.A., Beaudet A.L., Clayton-Smith J. et al. Angelman syndrome2005: updated consensus for diagnostic criteria. Am J Med Genet A. 2006;140(5):413-418. DOI: 10.1002/ajmg.a.31074
52. Lossie AC, Whitney MM, Amidon D, et al. Distinct phenotypes distinguish the molecular classes of Angelman syndromeJournal of Medical Genetics 2001;38:834-845
53. Poyatos D, Guitart M, Gabau E, et al. Severe phenotype in Angelman syndrome resulting from paternal isochromosome 15. J Med Genet. 2002 Feb;39(2):E4. doi: 10.1136/jmg.39.2.e4.
54. Varela, M., Kok, F., Otto, P. et al. Phenotypic variability in Angelman syndrome: comparison among different deletion classes and between deletion and UPD subjects. Eur J Hum Genet 12, 987–992 (2004). https://doi.org/10.1038/sj.ejhg.5201264
55. Shinji Saitoh, Takahito Wada, Maki Okajima, Kyoko Takano, Akira Sudo, Norio Niikawa. Uniparental disomy and imprinting defects in Japanese patients with Angelman syndrome. Brain Dev. 2005 Aug;27(5):389-91. doi: 10.1016/j.braindev.2003.12.013.
56. Luk HM, Lo IF. Angelman syndrome in Hong Kong Chinese: A 20 years' experience. Eur J Med Genet. 2016 Jun;59(6-7):315-9. doi: 10.1016/j.ejmg.2016.05.003. Epub 2016 May 9. PMID: 27174604
57. Hnoonual A, Kor-Anantakul P, Charalsawadi C, Worachotekamjorn J, Limprasert P. Case Report: An Atypical Angelman Syndrome Case With Obesity and Fulfilled Autism Spectrum Disorder Identified by Microarray. Front Genet. 2021 Sep 22;12:755605. doi: 10.3389/fgene.2021.755605. PMID: 34630535; PMCID: PMC8494305
58. Brennan ML, Adam MP, Seaver LH, Myers A, Schelley S, Zadeh N, Hudgins L, Bernstein JA. Increased body mass in infancy and early toddlerhood in Angelman syndrome patients with uniparental disomy and imprinting center defects. Am J Med Genet A. 2015 Jan;167A(1):142-6. doi: 10.1002/ajmg.a.36831. Epub 2014 Nov 17. PMID: 25402239.
59. Ioannides Y, Lokulo-Sodipe K, Mackay DJ, Davies JH, Temple IK. Temple syndrome: improving the recognition of an underdiagnosed chromosome 14 imprinting disorder: an analysis of 51 published cases. J Med Genet. 2014;51(8):495‐501.
60. Geoffron S, Abi Habib W, Chantot-Bastaraud S, et al. Chromosome 14q32.2 imprinted region disruption as an alternative molecular diagnosis of silver-Russell syndrome. J Clin Endocrinol Metab. 2018;103(7):2436‐2446.
61. Juriaans AF, Kerkhof GF, Mahabier EF, et al. Temple syndrome: clinical findings, body composition and cognition in 15 patients. J Clin Med. 2022;11(21):6289.
62. I K Temple, A Cockwell, T Hassold, D Pettay, P Jacobs. Maternal uniparental disomy for chromosome 14. J Med Genet. 1991 Aug;28(8):511–514. doi: 10.1136/jmg.28.8.511
63. Alicia F Juriaans, Gerthe F Kerkhof, Eva F Mahabier, Theo C J Sas, Nitash Zwaveling-Soonawala, Robbert N H Touwslager, Joost Rotteveel, Anita C S Hokken-Koelega. Temple Syndrome: Clinical Findings, Body Composition and Cognition in 15 Patients. J Clin Med. 2022 Oct 25;11(21):6289. doi: 10.3390/jcm11216289
64. Tomoe Ogawa, Hiromune Narusawa, Keisuke Nagasaki, Rika Kosaki, Yasuhiro Naiki, Michihiko Aramaki, Keiko Matsubara, Akie Nakamura, Maki Fukami, Tsutomu Ogata, Masayo Kagami. Temple Syndrome: Comprehensive Clinical Study in Genetically Confirmed 60 Japanese Patients. J Clin Endocrinol Metab. 2024 Dec 18:dgae883. doi: 10.1210/clinem/dgae883.
65. Béatrice Dubern, Héléna Mosbah, Marie Pigeyre, Karine Clément, Christine Poitou. Rare genetic causes of obesity: Diagnosis and management in clinical care. Ann Endocrinol (Paris). 2022 Feb;83(1):63-72. doi: 10.1016/j.ando.2021.12.003.
66. Takaesu Y., Komada Y., Inoue Y. Melatonin profile and its relation to circadian rhythm sleep disorders in Angelman syndrome patients. Sleep Med 2012; 13(9): 1164–1170. DOI: 10.1016/j.sleep.2012.06.015
67. Е. Г. Панченко, О. В. Васюкова, П. Л. Окороков, Д. А. Копытина, В. О. Сигин, В. В. Стрельников, Д. В. Залетаев. Клинический случай многолокусного нарушения импринтинга: первое описание в Российской Федерации. Российский вестник перинатологии и педиатрии. 2024;69(4):90-96. https://doi.org/10.21508/1027-4065-2024-69-4-90-96
68. Васюкова О.В., Окороков П.Л., Малиевский О.А., Неймарк А.Е., Зорин Е.А., Яшков Ю.И., Бурмицкая Ю.В., Копытина Д.А., Безлепкина О.Б., Петеркова В.А. Клинические рекомендации «Ожирение у детей». Ожирение и метаболизм. 2024;21(4):439-453. https://doi.org/10.14341/omet13194
69. Копытина Д.А., Васюкова О.В., Салахов Р.Р., Окороков П.Л., Копытина Е.В., Нагаева Е.В., Хусаинова Р.И., Минниахметов И.Р., Попов С.В., Безлепкина О.Б., Мокрышева Н.Г. Идентификация новых патогенных вариантов в гене GNAS у детей с морбидным ожирением и псевдогипопаратиреозом. Ожирение и метаболизм. 2024;21(4):412-424. https://doi.org/10.14341/omet13169
Васюкова Ольга Владимировна, к.м.н.
Researcher ID: AAO-3750-2020
Scopus Author ID: 57192194141
Москва
Авторы декларируют отсутствие конфликта интересов
Атавина-Ермакова Евгения Павловна
Москва
Авторы декларируют отсутствие конфликта интересов
Панченко Елизавета Григорьевна, м.н.с.
ID РИНЦ: 1080414
WoS: JXX-3224-2024
Москва
Авторы декларируют отсутствие конфликта интересов
Копытина Дарья Александровна
Scopus Author ID: 58853779500
Москва
Авторы декларируют отсутствие конфликта интересов
Симонова Ольга Анатольевна
Scopus: 56107313400
WoS: D-2157-2012
ID РИНЦ: 896923
Москва
Авторы декларируют отсутствие конфликта интересов
Удалова Василиса Юрьевна
Scopus ID: 57897149800
Москва
Авторы декларируют отсутствие конфликта интересов
Канивец Илья Вячеславович
Москва
Авторы декларируют отсутствие конфликта интересов
Акимова Дарья Борисовна
Scopus ID: 59341366000
WoS: ADV-3867-2022
Москва
Авторы декларируют отсутствие конфликта интересов
Скоблов Михаил Юрьевич, к.б.н.
РИНЦ Author ID: 124376
Scopus ID 8979068100
WoS: P-1387-2015
Москва
Авторы декларируют отсутствие конфликта интересов
Андреева Мария Александровна
Москва
Авторы декларируют отсутствие конфликта интересов
Нагаева Елена Витальевна, д.м.н.
Москва
Авторы декларируют отсутствие конфликта интересов
Мокрышева Наталья Георгиевна, д.м.н., профессор, академик РАН
ResearcherID: AAY-3761-2020
Scopus Author ID: 35269746000
Москва
Авторы декларируют отсутствие конфликта интересов
|
|
1. Рисунок 1. Частота БГИ в структуре исследуемой группы пациентов. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(310KB)
|
Метаданные ▾ | |
|
|
2. Рисунок 2. Частота осложнений, ассоциированных с ожирением и избыточной массой тела, у пациентов с БГИ. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(215KB)
|
Метаданные ▾ | |
|
|
3. Рисунок 3. Фенотипические особенности у девочки с синдромом Ангельмана: широкий, часто открытый рот, счастливый внешний вид, плосковальгусные стопы, морбидное ожирение. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(897KB)
|
Метаданные ▾ | |
|
|
4. Рисунок 4. Результаты МЧ-MLPA пациентки С (№140). Столбец «140—r-» — результат анализа копийности; референсный интервал: 0,80–1,20. Столбец «140—r+» — результат анализа метилирования; референсный интервал: 0,40–0,65. Выявлено гипометилирование SNRPN (2 зонда из 2). | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(2MB)
|
Метаданные ▾ | |
|
|
5. Рисунок 5. Молекулярно-генетический анализ пациента с нонсенс-вариантом c.2457T>A в гене MAGEL2. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(454KB)
|
Метаданные ▾ | |
Васюкова О.В., Атавина-Ермакова Е.П., Панченко Е.Г., Копытина Д.А., Симонова О.А., Удалова В.Ю., Канивец И.В., Акимова Д.Б., Скоблов М.Ю., Андреева М.А., Нагаева Е.В., Мокрышева Н.Г. Болезни геномного импринтинга в структуре синдромального ожирения у детей. Ожирение и метаболизм. 2025;22(3):180-195. https://doi.org/10.14341/omet13276
Vasyukova O.V., Atavina-Ermakova E.P., Panchenko E.G., Kopytina D.A., Simonova O.A., Udalova V.Y., Kanivets I.V., Akimova D.B., Skoblov M.Y., Andreeva M.A., Nagaeva E.V., Mokrysheva N.G. Genomic imprinting disorders in the structure of syndromic obesity in children. Obesity and metabolism. 2025;22(3):180-195. (In Russ.) https://doi.org/10.14341/omet13276
 |
117292
Россия, Москва, ул. Дм. Ульянова, д.11
____________________________________
117292
11, Dm. Ul’yanova str., Moscow, Russian Federation



































