




Contents
Scroll to:
https://doi.org/10.14341/omet12778
Scroll to:
Physiologically, autophagy is a major protective mechanism of β-cells from apoptosis, through can reserve normal β- cell mass and inhibit the progression of β-cells destruction. Beta-cell mass can be affected by differentiation from progenitors and de-differentiation as well as self-renewal and apoptosis. Shred evidence indicated that hypoglycemic drugs can induce β-cell proliferation capacity and neogenesis via autophagy stimulation. However, prolonged use of selective hypoglycemic drugs has induced pancreatitis besides several other factors that contribute to β-cell destruction and apoptosis initiation. Interestingly, some nonhypoglycemic medications possess the same effects on β-cells but depending on the combination of these drugs and the duration of exposure to β-cells. The paper comprehensively illustrates the role of the hypoglycemic drugs on the insulin-producing cells and the pathogeneses of β-cell destruction in type 2 diabetes mellitus, in addition to the regulation mechanisms of β-cells division in norm and pathology. The grasping of the hypoglycemic drug’s role in beta-cell is clinically crucial to evaluate novel therapeutic targets such as new signaling pathways. The present paper addresses a new strategy for diabetes mellitus management via targeting specific autophagy inducer factors (transcription factors, genes, lipid molecules, etc.).
Marzoog B.A., Vlasova T.I. Beta-cell autophagy under the scope of hypoglycemic drugs; possible mechanism as a novel therapeutic target. Obesity and metabolism. 2021;18(4):465-470. https://doi.org/10.14341/omet12778
Hypoglycemic drugs are extremely effective in relieving and controlling hyperglycemia, but unfortunately, recent data showed serious side effects after their administration [1]. Under normal physiological conditions, the balance between self-renewal and apoptosis maintains the β-cell mass at the familiar level. Whatever perturbance to this balance results in unfavorable outcomes. Diabetes mellitus usually arises on the background of metabolic syndrome that nowadays exponentially increases with the COVID-19 pandemic, especially in the caloric rich intake populations [2][3]. Shred evidence that dipeptidyl peptidase 4 (DPP4) can be a potential receptor for COVID-19 entrance, but administration of DPP4 inhibitors was not related to decreasing COVID-19 morbidity incidence [4]. Besides, the primary receptor for COVID-19, the angiotensin-converting enzyme receptor, is also expressed on pancreatic beta cells, therefore, COVID-19 has additional potential for beta-cell destruction and ketosis-prone diabetes developing [5]. Three main kinds of autophagy distinguished; macroautophagy, microauophagy, and chaperon mediated autophagy (we use autophagy term to describe macroautophagy in this paper). The paper aimed to analyze data from the present literature datadase (Scopus and Medline) on the effect of problem of the hypoglycemic drug’s effect on β-cell and their role in future therapeutic strategies. We searched both databases using keywords: «hypoglycemic drugs», «beta cell», «Metformin, Sulfonylureas», «Thiazolidinediones», «Sodium–glucose cotransporter 2 inhibitors», «Autophagy». 54 results in Scopus and 68 results in Medline. Excluding duplicates and not related titles to our paper by title remained 90 papers. Then we read the abstract of these papers, and only 47 papers related to our topic. After that, we read the full text of these 47 papers. The study of the effects of hypoglycemic drugs on β-cells has become an urgent problem in recent decades, as the number of candidates and affected individuals has increased dramatically.
The β-cells division is strictly controlled by variable factors in norm and pathology. A recent clinical study approved in vitro that β-cells can be differentiated into glucagon-producing cells through specific regulatory transcription factors [6]. Physiologically, the β-cell has a FOXO1 transcription factor (TF) in addition to the NKX6.1 transcription factor in the nucleus and cytoplasm [7][8]. The depletion of FOXO1 leads to the inability to retain NKX6.1 in the nucleus and consequently the deprivation of β-cells. At the same time, the β-cells have shown a capacity to transfer into progenitor-like state that have transcription factor Neurogenin3 that can give rise to any type of islets of Langerhans cells of similarity such as α and δ like cells in human type 2 diabetes [7][9][10]. Surprisingly, the study concluded that insulin secretion is inversely correlated with the degree of dedifferentiation, defined as the ratio of Syn-positive/hormone-negative cells to Syn-positive cells. On the other hand, β-cell differentiation does not depend on the age, weight or duration of diabetes [7]. Scientists believe that β-cells differentiation of -cells in type 2 diabetes is to protect them from apoptosis and immune system attack. Thereafter, when will be favorable metabolic conditions, the differentiated β-cells return into active β-cells. Proinflammatory cytokines such as IL-1β and IFN-γ were recently been shown to stimulate early phases of autophagy through ER stress-dependent activation of adenosine 5′monophosphate-activated protein kinase and the formation of reactive oxygen species formation [11]. In addition to inhibiting β-cell autophagic flux of cells due to impaired lysosomal function, which contributed to β-cell apoptosis [12]. Contrary, interleukin 22 (IL-22) and IL-6 have a cytoprotective effect on β-cells by stimulating autophagy and protecting β-cells from tumor necrosis factor-α, IL-1β, and interferon-γ induced apoptosis [13][14].
In the few past years, researchers had amplified the role of apoptosis, autophagy, oxidative stress, and nutrient overload in the regulation of insulin-producing cell activity and quantity [15][16]. Where Autophagy appears to play a significant role in the regulation of insulin homeostasis in addition to its role in β-cell survival [11][17]. Moreover, autophagy supports the proper differentiation of β-cells during development and contributing to β-cell function [18][19]. Studies in vitro have shown that autophagy protects the β-cells from apoptosis induced solely by fatty acids such as palmitate and cholesterol through activation of the Nuclear factor E2-related factor 2 (Nrf2) [20][21]. The Atg7 gene was found as an important regulator in autophagy deriving while Atg7 depletion results in beta mass decreasing, and glucose tolerance impairment, defective insulin secretion, and increased apoptosis when combined with high-fat and high-glucose diet, indicating Atg7 depletion impairs autophagy stimulation [22]. In contrast, autophagy can be stimulated by specific dietary components, GLP-1, and cytokines [17]. Excess lipid level plays a huge regulatory role in autophagy controlling too [20, 21]. On the other hand, few data suggested that starvation and amino acid deprivation can inhibit autophagy and promote crinophagy via direct fusing of insulin granules with the lysosomes [17, 23]. A single study published in Medicine journal indicated the role of microbiota in the regulation of homeostasis and eventually insulin receptors sensitivity that can be associated with type 2 diabetes mellitus appearance [24]. On the cellular level of regulation, the misbalance between fuel production by the mitochondria and its expenditure can be a positive stimulus to initiate insulin receptor resistance and accordingly type 2 diabetes mellitus [25]. Finally, autophagy plays a key role in driving the pancreatic stem cells (PSC) differentiation, animal models, into insulin-producing cells via an ambiguous mechanism suspected to be through the Wnt/B-catenin signaling pathway [19, 26].
Autophagy maintains beta cell homeostasis through redistribution of energy sources within the cell to preserve the most vital organelles from failure and later degradation [27]. Maintaining the anatomical structure of the beta cell requires autophagy, where the depletion of Atg7 in the beta cells results in impaired transmission activity of LC3-I to LC3-II transmission activity as well as accumulation of p62 and polyubiquitin in the beta cell cytoplasm, which is usually seen in diabetic patients [28]. Moreover, pathoanatomical changes were observed in Atg7 deficient beta cells such as cyst-like formation sized 15–20 mm that were associated with caspase-3-positive apoptotic cells.
Diabetes mellitus can arise under various pathological mechanisms that, in turn, culminate in the diminish of β-cell activity. Therefore, there should be different pathophysiological pathways for the development of the condition. Many of these mechanisms have been shown to have a genetic predisposition, including; Proopiomelanocortin gene mutation, Melanocortin receptor mutations, brain-derived neurotrophic factor and receptor mutations, glucose kinase mutations, hepatocyte nuclear factor mutations, mitochondrial DNA mutations, Insulin receptor mutations, viral oncogene homolog 2 (AKT2), v-akt murine thymoma and even lipodystrophy can alter the normal function of β-cell [29–33]. Apoptosis appears to be the leading cause of β-cell destruction in both types of diabetes mellitus through activation of interleukin (IL)-1β, nuclear factor (NF)-κB, and Fas pathways, through a process, known as insulitis [34]. The β-cells are highly sensitive to destructive pathogenic factors such as environmental regulated by physical activity, diet and intestinal microbiota [32]. High serum free fatty acid (FFA) and hyperglycemia are the primary cause of β-cells dysfunction via ER stress. And this in turn trigger NF-κB–dependent mechanism that culminates in caspase-3 activation for cytokines and an NF-κB–independent mechanism for nutrients “glucose hypersensitization” consequently apoptosis of β-cells [8][34][35]. In addition to decreasing in glucose-induced insulin secretion by β-cell due to failure of β-cell sensitivity to hyperglycemia and loss of the first phase with a decrease in the second phase of insulin secretion [36]. In most patients who suffer from type 2 diabetes, it is not so crucial the depletion in the mass of β-cells through dyslipidemia and hyperglycemia, via apoptosis [37][38]. Several studies stress the neurohormonal role in the pathogenesis of type 2 diabetes mellitus through the dysregulation of leptin hormone that leads to hyperphagia and obesity and accordingly insulin resistance [29]. IL-17 showed a piece of shred evidence in the pathophysiology of type 2 diabetes mellitus through enhancement of the inflammatory processes and insulin resistance where administration of IL-17 antagonist decreased the risk of type 2 diabetes mellitus emergence [39]. Recently, a study done by Anne Raimondo and her colleagues demonstrated that Peptidylglycine Alpha-amidating Monooxygenase (PAM) contribute to the development of type 2 diabetes mellitus [33].
Beta-cell dysfunction involved autophagy disturbance, where several studies reported that Atg7 knockdown cells suffered from impaired insulin secretion. Impaired autophagy induces the transdifferentiation of beta cells into alpha cells and non functional islet cells as well as impairs proliferation [40].
Many factors contribute to β-cell co-working ability such as hypoglycemic drugs. Therefore, various effects can be revealed on the administration of antihyperglycemic drugs depending on their mechanism of action. The drugs administered most frequently to control hyperglycemia are metformin and GLP-1 mimetics such as GLP-1 analogs and GLP-1-like molecules, exendin-4/exenatide, and its derivative such as lixisenatide [41]. Although all these drugs have shown a favorable effect on β-cells and reserving their mass and function. The clinical findings have shown that metformin promotes β-cells autophagy and poses an anti-inflammatory effect that enhances even immune response against viral infection when combined with the seasonal influenza vaccine [42–44]. Both groups of GLP-1 mimetics, GLP-1 receptor agonists and DPP-IV inhibitors, have been shown to induce autophagy in diabetic patients and enhance the insulin gene transcription and biosynthesis through the reduction in β-arrestin recruitment and faster agonist dissociation rates [45–47]. Indeed these two groups of medications have various effects on β-cells since they serve differently from each other [41]. The administration of GLP-1 agonists to normal and diabetic rodents has stimulated β-cells proliferation, neogenesis, and protects against apoptosis and inflammation [48–50]. While GLP-1 agonists long prescribing ended with inducing β–cell mass through the activation of multiple signaling pathways such as PKA, PI3-kinase, and ERK1/2 [51–53]. In particular, the liraglutide and exenatide administration in animal models promoted the first- and second-phase insulin secretion via restoring the beta-cell sensitivity to glucose, besides, the liraglutide and exenatide induced beta cell proliferation/regeneration and mass of beta cells through the protection from apoptosis [53, 54]. In vivo, few findings have shown that GLP-1R signaling exerts a vigorous effect on β-cell survival compared with DPP4 inhibition [55][56]. The prolonged administration of DPP4 inhibitors induced beta cells mass and regeneration capacity in addition to the anti-inflammatory response [57][58]. The inhibition of the mammalian target of rapamycin complex 1 (mTORC 1) has shown a stimulatory effect on the autophagy of β-cell, an example of such drugs is rapamycin [59][60]. In many clinical studies, this immunosuppressant and anti-neoplastic drug has lowered the glucose level and decreased weight gain in addition to insulin resistance [61–65]. However, unfortunately, several studies have shown that Rapamycin has impaired glucose tolerance and elevated insulin resistance even the appearance of frank diabetes in vivo in addition to a decrease in β-cell autophagy and function [62][66–70].
Sodium–glucose cotransporter 2 inhibitors (SGLT2) have beneficial effects on beta cells by inducing autophagy through the overexpression of adenosine monophosphate-activated protein kinase, sirtuin-1, and/or hypoxia-inducible factors-1α/2α. In vivo, SGLT2 has induced beta-cell proliferation and improved insulin secretion in response to hyperglycemia in addition to ameliorating lipotoxicity [71][72].
Sulfonylureas (SUs) is a common class of antihyperglycemic drug used in type 2 diabetes mellitus. Glibenclamide, second generation in this group of medications, induces beta cell autophagy by activating the AMPK pathway instead of the mTOR pathway However, the effects of autophagy depend on the state of the beta cells, if it is sensitive to hyperglycemia, thus enhancing autophagy induces insulin secretion. However, in altered beta cell sensitivity to high glucose levels, autophagy induction reduces insulin secretion. Also, it is well known that insulin over secretion induces endoplasmic reticulum stress and oxidative stress. Furthermore, over-induction of autophagy alters insulin storage granules and results in their degradation. studies on Min-6 cells suggested that Glibenclamide induces insulin secretion but its inhibition was accompanied by significant upregulation of insulin secretion [73].
Thiazolidinediones are another class of antihyperglycemic medication. Rosiglitazone belongs to this class of medications, which is reported to be a well inducer for beta cell autophagy through activation of the AMPK pathway in INS-1 cells. Beta cell autophagy maintains beta cells mass and prevent transdifferentiation (cell linage reprogramming) as well as improves proliferation.
To decrease the speed of β-cell destruction, it’s now recommended the early diagnosis of type 2 diabetes mellitus and prefers to not use insulin secretagogues such as sulfonylureas since they impair the β-cell function and leads to their apoptosis [66].
Finally, it seems that prolonged use of incretin-based therapies, particularly sitagliptin, for more than 2.4 years may promote the development of acute pancreatitis, especially when there is a tripling in the level of pancreatic amylase and lipase enzymes level [1][74]. (Figure 1)
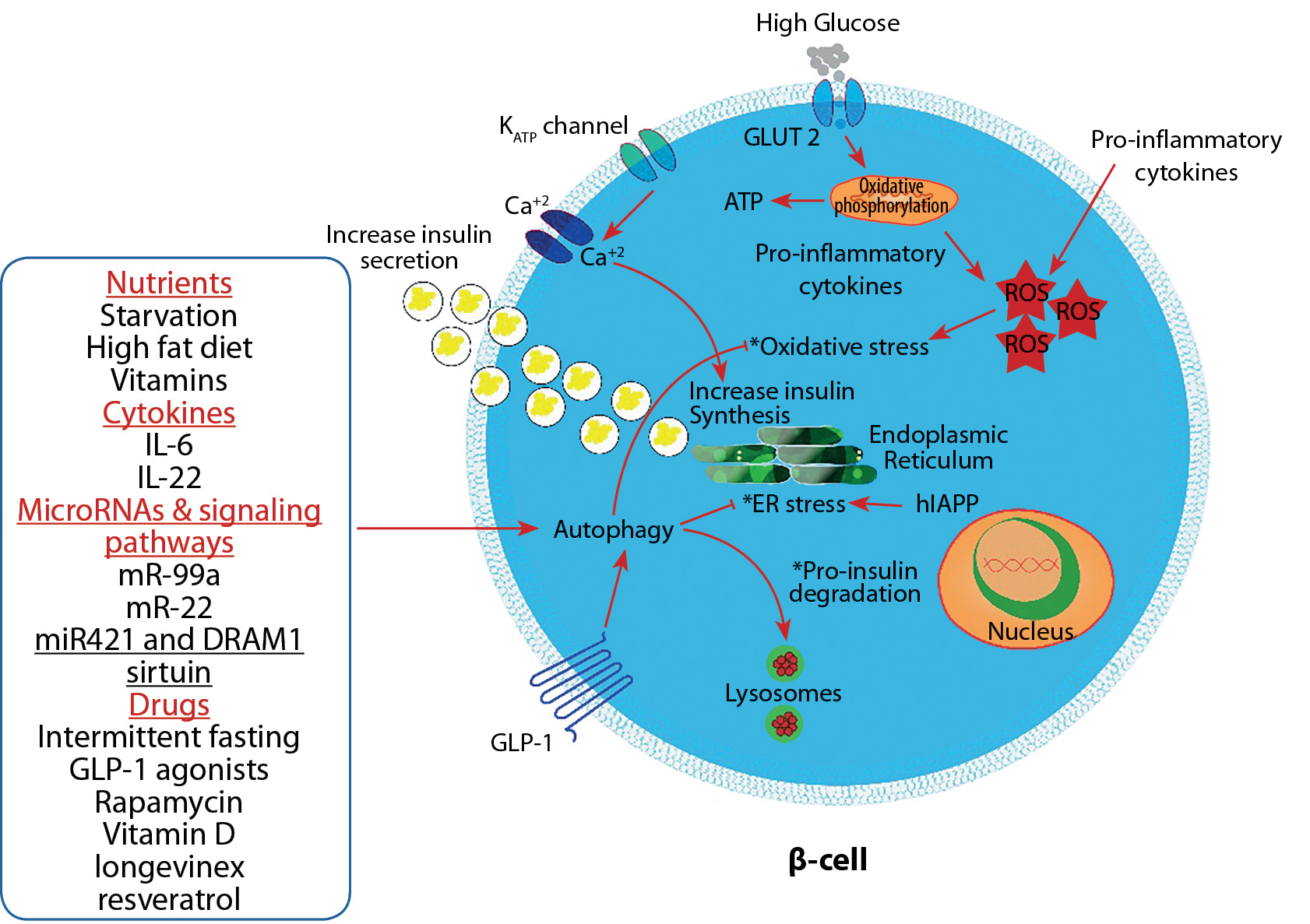
Figure 1. Autophagy’s role in maintaining Beta-cell mass and function through resolving the oxidative stress, endoplasmic reticulum (ER) stress, and reducing pro-insulin degradation (these functions indicated by asterisks).
Note. GLP-1 group of anti-diabetic medications in addition to other factors can affect autophagy. Oxidative stress and endoplasmic reticulum stress are critical for maintaining autophagy and beta cell mass from apoptosis and cell death, which are necessary to maintain blood glucose levels. Abbreviations: ATP — adenosine triphosphate, IL — interleukin, GLP-1 — glucagon-like peptide-1, ROS — reactive oxygen species, hIAPP — human islet amyloid polypeptide.
Therefore, the observed data have shown the possibility of β-cell mass maintaining in diabetic patients by promoting autophagy promotion via selective hypoglycemic drugs, including DDP-IV inhibitors and GLP-1 mimetics. Several preclinical trials have been performed in the course of stimulating β-cell proliferation by using hypoglycemic drugs. Most were promising and culminated in the possibility of β-cell to maintain self-renewal and protect themselves from transdifferentiation into pathological nonbeta cells. But, this process indirectly occurs, firstly hypoglycemic drugs induce autophagy, then autophagy promoted beta-cell proliferation, therefore there is no direct in vitro or in vivo study shown this, and we in this paper connected the missing parts to complete the chain [7][17][19][42][43][73][75–77].
In preclinical studies, the majority of hypoglycemic drugs possessed therapeutic effects on the beta cell during their administration including; enhance autophagy and insulin biosynthesis, possessing anti-inflammatory effects, protecting against apoptosis and beta cell destruction, as well as enhance β-cell survival. Beta-cell mass can be affected by differentiation from progenitors and de-differentiation as well as self-renewal and apoptosis.
Interestingly, recent investigated data have emphasized the role of transdifferentiation in beta cell regeneration by recruiting endoderm-derived cells by applying specific niche factors, including transcription factors [78–80]. But, the following question is still ambiguous and needs more research and clinical study; Do neo transdifferentiated beta cells persist or terminate with stop of the administration of the inducing factor administration? While this can be of use in triggering other cells of islets of Langerhans to transfer into β-cells and start producing insulin. This requires more research and clinical studies to prove the clinical importance of the hypoglycemic drugs on β-cells metabolism. We suggest inducing beta cells autophagy in diabetic patients through targeting autophagy signaling pathways ameliorates hyperglycemia. Hypoglycemic drags are well inducers for beta-cells autophagy and can be recruited and modified to be suitable for this purpose.
Funding. No funding.
Conflict of interest. The authors declare no obvious and potential conflicts of interest related to the content of this article.
Contribution of authors. Basheer Abdullah Marzoog — design of the work and the acquisition, analysis, interpretation of data for the work, Drafting the work and revising it critically for important intellectual content. Tatyana Ivanovna Vlasova — revising the work critically for important intellectual content. All of the authors read and approved the final version of the manuscript before publication, agreed to be responsible for all aspects of the work, implying proper examination and resolution of issues relating to the accuracy or integrity of any part of the work.
1. DeVries JH, Rosenstock J. DPP-4 Inhibitor-Related Pancreatitis: Rare but Real! Diabetes Care. 2017;40(2):161-163. doi:10.2337/dci16-0035
2. Sada K, Nishikawa T, Kukidome D, et al. Hyperglycemia induces cellular hypoxia through production of mitochondrial ROS followed by suppression of aquaporin-1. PLoS One. 2016;11(7). doi:10.1371/journal.pone.0158619
3. Lim S, Bae JH, Kwon H-S, Nauck MA. COVID-19 and diabetes mellitus: from pathophysiology to clinical management. Nat Rev Endocrinol. 2021;17(1):11-30. doi:10.1038/s41574-020-00435-4
4. Rubino F, Amiel SA, Zimmet P, et al. New-Onset Diabetes in Covid-19. N Engl J Med. Published online 2020. doi:10.1056/nejmc2018688
5. Brereton MF, Iberl M, Shimomura K, et al. Reversible changes in pancreatic islet structure and function produced by elevated blood glucose. Nat Commun. 2014;5(1):4639. doi:10.1038/ncomms5639
6. Cinti F, Bouchi R, Kim-Muller JY, et al. Evidence of β-Cell Dedifferentiation in Human Type 2 Diabetes. J Clin Endocrinol Metab. 2016;101(3):1044-1054. doi:10.1210/jc.2015-2860
7. Cheng STW, Li SYT, Leung PS. Fibroblast Growth Factor 21 Stimulates Pancreatic Islet Autophagy via Inhibition of AMPK-mTOR Signaling. Int J Mol Sci. 2019;20(10):2517. doi:10.3390/ijms20102517
8. Talchai C, Xuan S, Lin H V., Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell. 2012;150(6):1223-1234. doi:10.1016/j.cell.2012.07.029
9. Bensellam M, Jonas JC, Laybutt DR. Mechanisms of β;-cell dedifferentiation in diabetes: Recent findings and future research directions. J Endocrinol. 2018;236(2):R109-R143. doi:10.1530/JOE-17-0516
10. DiNicolantonio JJ, McCarty M. Autophagy-induced degradation of Notch1, achieved through intermittent fasting, may promote beta cell neogenesis: implications for reversal of type 2 diabetes. Open Hear. 2019;6(1):e001028. doi:10.1136/openhrt-2019-001028
11. Lambelet M, Terra LF, Fukaya M, et al. Dysfunctional autophagy following exposure to pro-inflammatory cytokines contributes to pancreatic β-cell apoptosis. Cell Death Dis. 2018;9(2):96. doi:10.1038/s41419-017-0121-5
12. Hu M, Yang S, Yang L, Cheng Y, Zhang H. Interleukin-22 alleviated palmitate-induced endoplasmic reticulum stress in INS-1 cells through activation of autophagy. PLoS One. 2016;11(1). doi:10.1371/journal.pone.0146818
13. Linnemann AK, Blumer J, Marasco MR, et al. Interleukin 6 protects pancreatic b cells from apoptosis by stimulation of autophagy. FASEB J. 2017;31(9):4140-4152. doi:10.1096/fj.201700061RR
14. Butler PC, Meier JJ, Butler AE, Bhushan A. The replication of β cells in normal physiology, in disease and for therapy. Nat Clin Pract Endocrinol Metab. 2007;3(11):758-768. doi:10.1038/ncpendmet0647
15. Talchai C, Lin HV, Kitamura T AD. Genetic and biochemical pathways of -cell failure in type 2 diabetes. Diabetes Obes Metab. 2009;11(suppl.4):38 – 45.
16. Marasco MR, Linnemann AK. B-Cell autophagy in diabetes pathogenesis. Endocrinology. 2018;159(5):2127-2141. doi:10.1210/en.2017-03273
17. Riahi Y, Wikstrom JD, Bachar-Wikstrom E, et al. Autophagy is a major regulator of beta cell insulin homeostasis. Diabetologia. 2016;59(7):1480-1491. doi:10.1007/s00125-016-3868-9
18. Ren L, Yang H, Cui Y, et al. Autophagy is essential for the differentiation of porcine PSCs into insulin-producing cells. Biochem Biophys Res Commun. 2017;488(3):471-476. doi:10.1016/j.bbrc.2017.05.058
19. Choi SE, Lee SM, Lee YJ, et al. Protective role of autophagy in palmitate-induced INS-1 β-cell death. Endocrinology. 2009;150(1):126-134. doi:10.1210/en.2008-0483
20. Wu J, Kong F, Pan Q, et al. Autophagy protects against cholesterol-induced apoptosis in pancreatic β-cells. Biochem Biophys Res Commun. 2017;482(4):678-685. doi:10.1016/j.bbrc.2016.11.093
21. Sheng Q, Xiao X, Prasadan K, et al. Autophagy protects pancreatic beta cell mass and function in the setting of a high-fat and high-glucose diet. Sci Rep. Published online 2017. doi:10.1038/s41598-017-16485-0
22. Goginashvili A, Zhang Z, Erbs E, et al. Insulin secretory granules control autophagy in Pancreatic β cells. Science (80- ). 2015;347(6224):878-882. doi:10.1126/science.aaa2628
23. Li C, Li X, Han H, et al. Effect of probiotics on metabolic profiles in type 2 diabetes mellitus. Med (United States). Published online 2016. doi:10.1097/MD.0000000000004088
24. Patti ME, Corvera S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr Rev. Published online 2010. doi:10.1210/er.2009-0027
25. Xu S, Sun F, Ren L, Yang H, Tian N, Peng S. Resveratrol controlled the fate of porcine pancreatic stem cells through the Wnt/β-catenin signaling pathway mediated by Sirt1. PLoS One. 2017;12(10). doi:10.1371/journal.pone.0187159
26. Murphy R, Carroll RW, Krebs JD. Pathogenesis of the metabolic syndrome: insights from monogenic disorders. Mediators Inflamm. 2013;2013:920214. doi:10.1155/2013/920214
27. Nica AC, Ongen H, Irminger J-C, et al. Cell-type, allelic, and genetic signatures in the human pancreatic beta cell transcriptome. Genome Res. 2013;23(9):1554-1562. doi:10.1101/gr.150706.112
28. Brunetti A, Chiefari E, Foti D. [Perspectives on the contribution of genetics to the pathogenesis of type 2 diabetes mellitus]. Recenti Prog Med. 2011;102(12):468-475. doi:10.1701/998.10858
29. Kalin MF, Goncalves M, John-Kalarickal J, Fonseca V. Pathogenesis of type 2 diabetes mellitus. In: Principles of Diabetes Mellitus: Third Edition. ; 2017. doi:10.1007/978-3-319-18741-9_13
30. Raimondo A, Thomsen SK, Hastoy B, et al. Type 2 Diabetes Risk Alleles Reveal a Role for Peptidylglycine Alpha-Amidating Monooxygenase in Beta Cell Function. bioRxiv; 2017:158642. doi:10.1101/158642
31. Cnop M, Welsh N, Jonas JC, Jörns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic β-cell death in type 1 and type 2 diabetes: Many differences, few similarities. Diabetes. 2005;54(SUPPL. 2):S97-S107. doi:10.2337/diabetes.54.suppl_2.S97
32. Ozougwu O. The pathogenesis and pathophysiology of type 1 and type 2 diabetes mellitus. J Physiol Pathophysiol. 2013;4(4):46-57. doi:10.5897/JPAP2013.0001
33. Ashcroft FM, Rorsman P. Molecular defects in insulin secretion in type-2 diabetes. Rev Endocr Metab Disord. 2004;5(2):135-142. doi:10.1023/B:REMD.0000021435.87776.a7
34. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52(1):102-110. doi:10.2337/diabetes.52.1.102
35. Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia. 2003;46(1):3-19. doi:10.1007/s00125-002-1009-0
36. Abdel-Moneim A, Bakery HH, Allam G. The potential pathogenic role of IL-17/Th17 cells in both type 1 and type 2 diabetes mellitus. Biomed Pharmacother. 2018;101:287-292. doi:10.1016/j.biopha.2018.02.103
37. Dalle S, Burcelin R, Gourdy P. Specific actions of GLP-1 receptor agonists and DPP4 inhibitors for the treatment of pancreatic β-cell impairments in type 2 diabetes. Cell Signal. 2013;25(2):570-579. doi:10.1016/j.cellsig.2012.11.009
38. Jiang Y, Huang W, Wang J, et al. Metformin Plays a Dual Role in MIN6 Pancreatic β Cell Function through AMPK-dependent Autophagy. Int J Biol Sci. 2014;10(3):268-277. doi:10.7150/ijbs.7929
39. Wu J, Wu JJ, Yang LJ, Wei LX, Zou DJ. Rosiglitazone protects against palmitate-induced pancreatic beta-cell death by activation of autophagy via 5′-AMP-activated protein kinase modulation. Endocrine. 2013;44(1):87-98. doi:10.1007/s12020-012-9826-5
40. Diaz A, Romero M, Vazquez T, Lechner S, Blomberg BB, Frasca D. Metformin improves in vivo and in vitro B cell function in individuals with obesity and Type-2 Diabetes. Vaccine. 2017;35(20):2694-2700. doi:10.1016/j.vaccine.2017.03.078
41. Janzen KM, Steuber TD, Nisly SA. GLP-1 Agonists in Type 1 Diabetes Mellitus. Ann Pharmacother. 2016;50(8):656-665. doi:10.1177/1060028016651279
42. Wajchenberg BL. β-cell failure in diabetes and preservation by clinical treatment. Endocr Rev. Published online 2007. doi:10.1210/10.1210/er.2006-0038
43. Jones B, Buenaventura T, Kanda N, et al. Targeting GLP-1 receptor trafficking to improve agonist efficacy. Nat Commun. Published online 2018. doi:10.1038/s41467-018-03941-2
44. Donnelly D. The structure and function of the glucagon-like peptide-1 receptor and its ligands. Br J Pharmacol. 2012;166(1):27-41. doi:10.1111/j.1476-5381.2011.01687.x
45. Shyangdan DS, Royle P, Clar C, Sharma P, Waugh N, Snaith A. Glucagon-like peptide analogues for type 2 diabetes mellitus. Cochrane Database Syst Rev. Published online 2011. doi:10.1002/14651858.CD006423.pub2
46. Piya MK, Tahrani AA, Barnett AH. Emerging treatment options for type 2 diabetes. Br J Clin Pharmacol. Published online 2010. doi:10.1111/j.1365-2125.2010.03711.x
47. Doyle ME, Egan JM. Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacol Ther. 2007;113(3):546-593. doi:10.1016/j.pharmthera.2006.11.007
48. Lee Y-S, Jun H-S. Anti-Inflammatory Effects of GLP-1-Based Therapies beyond Glucose Control. Mediators Inflamm. 2016;2016:1-11. doi:10.1155/2016/3094642
49. Lee YS, Jun HS. Anti-diabetic actions of glucagon-like peptide-1 on pancreatic beta-cells. Metabolism. Published online 2014. doi:10.1016/j.metabol.2013.09.010
50. Vilsbøll T. The effects of glucagon-like peptide-1 on the beta cell. Diabetes, Obes Metab. Published online 2009. doi:10.1111/j.1463-1326.2009.01073.x
51. Pratley RE, Nauck M, Bailey T, et al. Liraglutide versus sitagliptin for patients with type 2 diabetes who did not have adequate glycaemic control with metformin: a 26-week, randomised, parallel-group, open-label trial. Lancet. 2010;375(9724):1447-1456. doi:10.1016/S0140-6736(10)60307-8
52. Bergenstal RM, Wysham C, MacConell L, et al. Efficacy and safety of exenatide once weekly versus sitagliptin or pioglitazone as an adjunct to metformin for treatment of type 2 diabetes (DURATION-2): A randomised trial. Lancet. 2010;376(9739):431-439. doi:10.1016/S0140-6736(10)60590-9
53. Omar BA, Vikman J, Winzell MS, et al. Enhanced beta cell function and anti-inflammatory effect after chronic treatment with the dipeptidyl peptidase-4 inhibitor vildagliptin in an advanced-aged diet-induced obesity mouse model. Diabetologia. Published online 2013. doi:10.1007/s00125-013-2927-8
54. Yang L, Yuan J, Zhou Z. Emerging Roles of Dipeptidyl Peptidase 4 Inhibitors: Anti-Inflammatory and Immunomodulatory Effect and Its Application in Diabetes Mellitus. Can J Diabetes. Published online 2014. doi:10.1016/j.jcjd.2014.01.008
55. Tanemura M, Ohmura Y, Deguchi T, et al. Rapamycin causes upregulation of autophagy and impairs islets function both in vitro and in vivo. Am J Transplant. 2012;12(1):102-114. doi:10.1111/j.1600-6143.2011.03771.x
56. Zhou Z, Wu S, Li X, Xue Z, Tong J. Rapamycin induces autophagy and exacerbates metabolism associated complications in a mouse model of type 1 diabetes. Indian J Exp Biol. 2010;48(1):31-38. http://www.ncbi.nlm.nih.gov/pubmed/20358864
57. Chang G-R, Wu Y-Y, Chiu Y-S, et al. Long-term administration of rapamycin reduces adiposity, but impairs glucose tolerance in high-fat diet-fed KK/HlJ mice. Basic Clin Pharmacol Toxicol. 2009;105(3):188-198. doi:10.1111/j.1742-7843.2009.00427.x
58. Chang G-R, Chiu Y-S, Wu Y-Y, et al. Rapamycin protects against high fat diet-induced obesity in C57BL/6J mice. J Pharmacol Sci. 2009;109(4):496-503. doi:10.1254/jphs.08215fp
59. Gong F-H, Ye Y-N, Li J-M, Zhao H-Y, Li X-K. Rapamycin-ameliorated diabetic symptoms involved in increasing adiponectin expression in diabetic mice on a high-fat diet. Kaohsiung J Med Sci. 2017;33(7):321-326. doi:10.1016/j.kjms.2017.05.008
60. Reifsnyder PC, Flurkey K, Te A, Harrison DE. Rapamycin treatment benefits glucose metabolism in mouse models of type 2 diabetes. Aging (Albany NY). 2016;8(11):3120-3130. doi:10.18632/aging.101117
61. Fang Y, Westbrook R, Hill C, et al. Duration of rapamycin treatment has differential effects on metabolism in mice. Cell Metab. 2013;17(3):456-462. doi:10.1016/j.cmet.2013.02.008
62. Lupi R, Del Prato S. Beta-cell apoptosis in type 2 diabetes: quantitative and functional consequences. Diabetes Metab. 2008;34 Suppl 2(SUPPL. 2):S56-64. doi:10.1016/S1262-3636(08)73396-2
63. Barlow AD, Nicholson ML, Herbert TP. Evidence for Rapamycin Toxicity in Pancreatic β-Cells and a Review of the Underlying Molecular Mechanisms. Diabetes. 2013;62(8):2674-2682. doi:10.2337/db13-0106
64. Schindler CE, Partap U, Patchen BK, Swoap SJ. Chronic rapamycin treatment causes diabetes in male mice. Am J Physiol Regul Integr Comp Physiol. 2014;307(4):R434-43. doi:10.1152/ajpregu.00123.2014
65. Lamming DW, Ye L, Astle CM, Baur JA, Sabatini DM, Harrison DE. Young and old genetically heterogeneous HET3 mice on a rapamycin diet are glucose intolerant but insulin sensitive. Aging Cell. 2013;12(4):712-718. doi:10.1111/acel.12097
66. Lamming DW, Ye L, Katajisto P, et al. Rapamycin-Induced Insulin Resistance Is Mediated by mTORC2 Loss and Uncoupled from Longevity. Science (80- ). 2012;335(6076):1638-1643. doi:10.1126/science.1215135
67. Ganesan K, Rana MBM, Sultan S. Oral Hypoglycemic Medications. StatPearls Publishing; 2020. Accessed November 12, 2020. http://www.ncbi.nlm.nih.gov/pubmed/29494008
68. Zhou J, Kang X, Luo Y, et al. Glibenclamide-Induced Autophagy Inhibits Its Insulin Secretion-Improving Function in β Cells. Int J Endocrinol. 2019;2019:1-8. doi:10.1155/2019/1265175
69. Bugliani M, Mossuto S, Grano F, et al. Modulation of Autophagy Influences the Function and Survival of Human Pancreatic Beta Cells Under Endoplasmic Reticulum Stress Conditions and in Type 2 Diabetes. Front Endocrinol (Lausanne). 2019;10. doi:10.3389/fendo.2019.00052
70. Chen Z, Li Y-B, Han J, et al. The double-edged effect of autophagy in pancreatic beta cells and diabetes. Autophagy. 2011;7(1):12-16. doi:10.4161/auto.7.1.13607
71. Capozzi ME, DiMarchi RD, Tschöp MH, Finan B, Campbell JE. Targeting the Incretin/Glucagon System with Triagonists to Treat Diabetes. Vol 39. Oxford University Press; 2018:719-738. doi:10.1210/er.2018-00117
72. Churchill AJ, Gutiérrez GD, Singer RA, Lorberbaum DS, Fischer KA, Sussel L. Genetic evidence that Nkx2.2 acts primarily downstream of Neurog3 in pancreatic endocrine lineage development. Elife. 2017;6:e20010. doi:10.7554/eLife.20010
73. Zhu Y, Liu Q, Zhou Z, Ikeda Y. PDX1, Neurogenin-3, and MAFA: Critical transcription regulators for beta cell development and regeneration. Stem Cell Res Ther. 2017;8(1):240. doi:10.1186/s13287-017-0694-z
74. Donelan W, Li S, Wang H, et al. Pancreatic and duodenal homeobox gene 1 (Pdx1) down-regulates hepatic transcription factor 1 alpha (hnf1α) expression during reprogramming of human hepatic cells into insulin-producing cells. Am J Transl Res. Published online 2015.
Basheer Abdullah Marzoog - undergraduate student; Researcher ID: AAD-6284-2021.
68 Bolshevitskaya str., 430005 Saransk
The authors declare no obvious and potential conflicts of interest related to the content of this article
Tatyana Ivanovna Vlasova, MD, PhD, professor; eLibrary SPIN: 5314-3771.
Saransk
The authors declare no obvious and potential conflicts of interest related to the content of this article
|
|
1. Figure 1. Autophagy’s role in maintaining Beta-cell mass and function through resolving the oxidative stress, endoplasmic reticulum (ER) stress, and reducing pro-insulin degradation (these functions indicated by asterisks). Note. GLP-1 group of anti-diabetic medications in addition to other factors can affect autophagy. Oxidative stress and endoplasmic reticulum stress are critical for maintaining autophagy and beta cell mass from apoptosis and cell death, which are necessary to maintain blood glucose levels. Abbreviations: ATP — adenosine triphosphate, IL — interleukin, GLP-1 — glucagon-like peptide-1, ROS — reactive oxygen species, hIAPP — human islet amyloid polypeptide. | |
| Subject | ||
| Type | Исследовательские инструменты | |
View
(390KB)
|
Indexing metadata ▾ | |
Marzoog B.A., Vlasova T.I. Beta-cell autophagy under the scope of hypoglycemic drugs; possible mechanism as a novel therapeutic target. Obesity and metabolism. 2021;18(4):465-470. https://doi.org/10.14341/omet12778
 |
117292
Россия, Москва, ул. Дм. Ульянова, д.11
____________________________________
117292
11, Dm. Ul’yanova str., Moscow, Russian Federation



































