



Содержание
Перейти к:
 М. А. Перепелова,
В. К. Слоква,
Е. А. Пигарова,
А. С. Шутова,
А. А. Колодкина,
А. В. Перепелов,
Л. К. Дзеранова
М. А. Перепелова,
В. К. Слоква,
Е. А. Пигарова,
А. С. Шутова,
А. А. Колодкина,
А. В. Перепелов,
Л. К. Дзеранова https://doi.org/10.14341/omet13287
Перейти к:
Гормоны щитовидной железы участвуют в активации гликогенолиза и митохондриального окислительного фосфорилирования. Синдром Кохера-Дебре-Семелена (СКДС), или по-другому «гипотиреоидная миопатия» характеризуется снижением гликогенолитической активности, что приводит к отложению гликогена в мышцах, запасы которого начинают истощаться по мере достижения эутиреоза.
В основном пациенты предъявляют жалобы на мышечную слабость, преимущественно проксимальных групп мышц, скованность, миалгию и судороги. Отличительной чертой гипотиреоидных миопатий является обратимость клинических проявлений и значительное улучшение самочувствия на фоне медикаментозной компенсации заболевания. Однако отсутствие яркой клинической картины гипотиреоза в сочетании с редкой встречаемостью затрудняет раннюю диагностику и часто требует дифференциального диагноза с другими видами миопатий. Представлен клинический случай сочетания генетически детерминированной миопатии с экстрапирамидными симптомами, ассоциированной с мутацией в гене MICU1 и гипотиреоидной миопатии на фоне врожденного гипотиреоза у пациента от близкородственного брака.
Перепелова М.А., Слоква В.К., Пигарова Е.А., Шутова А.С., Колодкина А.А., Перепелов А.В., Дзеранова Л.К. Гипотиреоидная миопатия и ее связь с мутациями в гене MICU1: клинический случай. Ожирение и метаболизм. 2025;22(3):263-268. https://doi.org/10.14341/omet13287
Perepelova M.A., Slokva V.K., Pigarova E.A., Shutova A.S., Kolodkina A.A., Perepelov A.V., Dzeranova L.K. Hypothyroid myopathy and its association with MICU1 gene mutations: a clinical caseauthors. Obesity and metabolism. 2025;22(3):263-268. (In Russ.) https://doi.org/10.14341/omet13287
Гипотиреоз — распространенное эндокринное заболевание щитовидной железы, которым страдают до 5% населения, однако из-за отсутствия специфических симптомов и сходства проявлений со многими психическими и соматическими болезнями его диагностика может представлять значительные трудности и нередко бывает несвоевременной, что ухудшает качество жизни пациентов [1].
Одним из наиболее частых осложнений гипотиреоза является поражение нервно-мышечной системы. Гипотиреоидная миопатия составляет около 5% всех приобретенных миопатий, а среди пациентов с первичным гипотиреозом этот синдром встречается с частотой от 25 до 60% [2]. Мышечные симптомы (скованность, миалгии, судороги, легкая утомляемость) отмечают большинство пациентов с гипотиреозом. Часто недооценивается тот факт, что мышечные симптомы могут быть преобладающим или единственным клиническим проявлением гипотиреоза, что ставит вопрос о дифференциальной диагностике с другими причинами миопатии. Отличительной чертой гипотиреоидной миопатии (включая полимиозитоподобный синдром) является полное клиническое выздоровление и разрешение лабораторных отклонений после назначения заместительной терапии гормонами щитовидной железы [3][4][5].
В ФГБУ «НМИЦ эндокринологии» Минздрава России поступил мужчина 26 лет с жалобами на выраженную общую слабость, сонливость, отсутствие аппетита, хронические запоры до 10 дней. В последние 1,5 года стала беспокоить прогрессирующая боль и слабость в мышцах нижних конечностей, нарушение координации, при поступлении передвигался с поддержкой, были трудности при самостоятельном передвижении.
Из анамнеза известно, что пациент — от близкородственного брака двоюродных брата и сестры. Первый из четырех детей в семье, у сибсов подобных отклонений не наблюдается.
В 13–14 лет (2009–2010 г.) отмечалось замедление темпов роста и развития. В 2014 г. (17 лет) диагностирован гипотиреоз, гипофизарный нанизм, по результатам магнитно-резонансной томографии (МРТ) выявлена аденома гипофиза. У специалистов не наблюдался. Через два года повторно обследован: пролактин — 402 мМЕ/л (90—350), соматотропный гормон (СТГ) — 6,67 МЕ/л (0,4–4), тиреотропный гормон (ТТГ) — 45,7 мМЕ/л (0,4–6). По результатам МРТ — данных о наличии объемных образований не получено. У затылочных рогов боковых желудочков нерезко выраженные очаги глиозного изменения вероятно воспалительного характера. Умеренно выраженная наружная заместительная гидроцефалия. Симптом «пустого» турецкого седла. По результатам ультразвукового исследования (УЗИ) щитовидной железы — гипоплазия правой доли и узловой зоб левой доли (образование с мелкокистозными включениями размером 1,5*1,6 см). Инициирована терапия левотироксином натрия, на фоне которого отмечалась компенсация гипотиреоза ТТГ — 4,52 мМЕ/л (0,4–6). В этот же период проводился курс инъекций гормона роста со слабой положительной динамикой. По данным рентгенографии в 20 лет, зоны роста полностью закрыты, препарат отменен.
В 2019 г. (23 года) обратился к андрологу с целью реализации репродуктивной функции. Назначена заместительная гормональная терапия (ЗГТ) препаратами тестостерона, после рождения здорового сына через год прием самостоятельно отменил.
В 26 лет пациент консультирован неврологом в ФГБУ «НЦН», выставлен предварительный диагноз полинейропатии, назначено дообследование. Проведена электронейромиография, на которой выявлены признаки генерализованных сенсорных невритических нарушений аксонального типа, более выраженных в нижних конечностях.
По данным физикального обследования: масса тела — 37,0 кг, рост — 143,5 см, индекс массы тела (ИМТ) — 18,0 кг/м² (рис. 1), бледность кожных покровов, одутловатость лица, параорбитальный отек, язык увеличен. Телосложение диспропорциональное, пропорции тела хондродистрофического типа, коэффициент «верхний сегмент/нижний сегмент» = 1,3:1 (норма для взрослого 1,1–0,9:1), что, вероятно, связано с нарушением эпифизарного роста костей. Отмечалась псевдогипертрофия мышц пояса верхних и нижних конечностей, мышечная сила диффузно снижена до четырех баллов, тонус симметрично повышен, сухожильно-периостальные рефлексы высокие, с расширением рефлексогенных зон. Движения в крупных суставах ограничены за счет мышечных контрактур. Походка с элементами атаксии, скованная, при передвижении держится за руку. Щитовидная железа не увеличена, безболезненна, узловые образования не пальпируются. У пациента отмечалось скудное оволосение лица, подмышечных впадин, паховой области (Tanner Р2), половые органы развиты соответственно возрасту (Tanner G5).
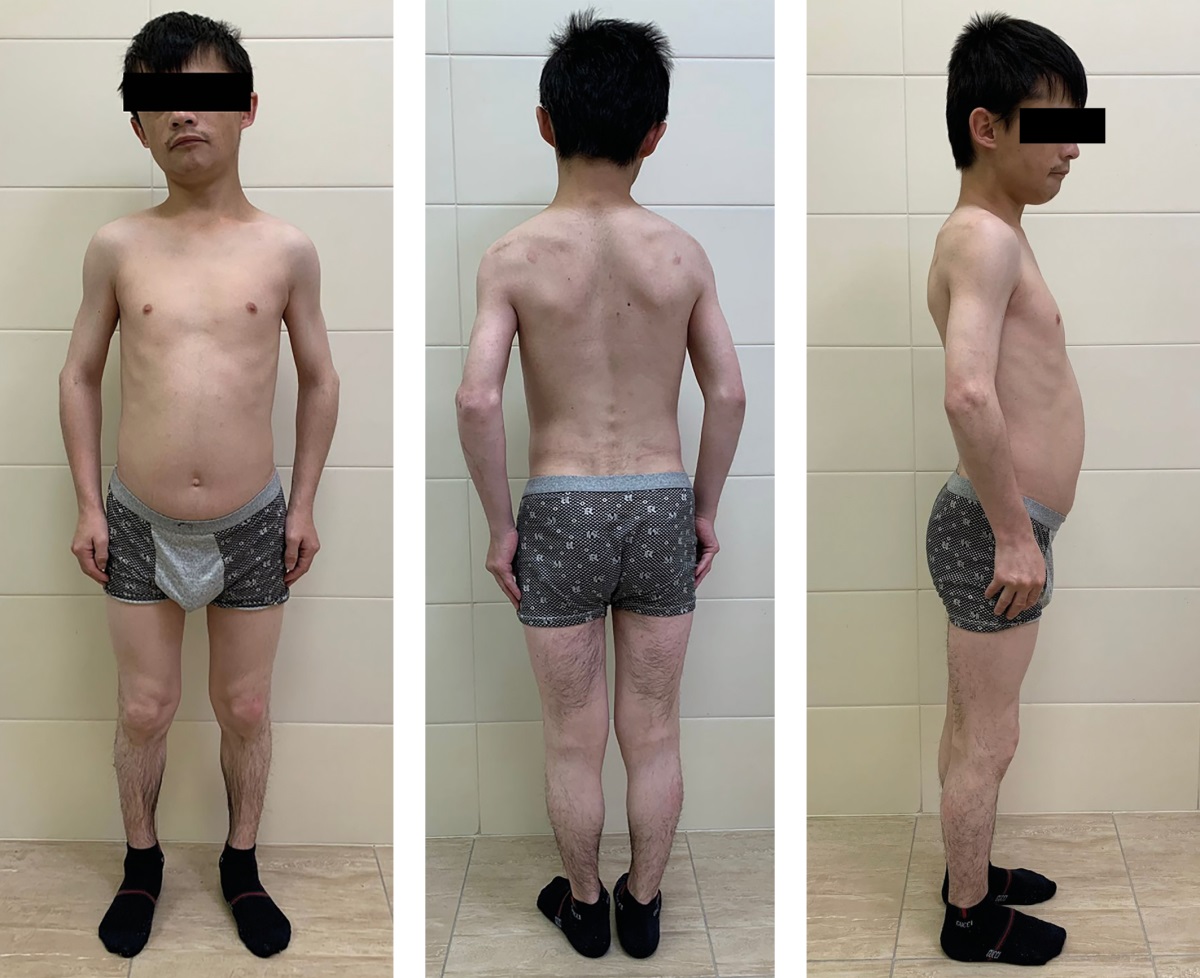
Рисунок 1. Пациент Э., 26 лет.
При гормональном анализе крови обнаружено значительное повышение уровня ТТГ — более 500 мМЕ/л (0,25–3,5), при разведении — 836 мМЕ/л, из которого биоактивный ТТГ — 174 мМЕ/л; Т4 свободный (свТ4) — 5,15 пмоль/л (9–19), Т3 свободный (свТ3) — 2,3 пмоль/л (2,6–5,7) — полученные данные расценены как первичный гипотиреоз в стадии длительной декомпенсации. Кроме того, у пациента диагностирован нормогонадотропный гипогонадизм: тестостерон — 5,98 нмоль/л (11–28,2), ЛГ — 3,43 Ед/л (2,5–11), ФСГ — 4,88 Ед/л (1,6–9,7), гиперпролактинемия — 753,9 мЕд/л (66–436), снижение ИФР-1 — 71,77 нг/мл (82–283), вероятнее всего, обусловленные гипотиреозом.
По результатам УЗИ щитовидной железы, где общий V=3,2 см³, структура ложа полностью представлена гиперэхогенной фиброзной тканью, в левой доле визуализируется образование размерами 1,6х1,4х0,9 см (EU-TIRADS 4). Выполнена тонкоигольная аспирационная биопсия, при цитологическом исследовании пунктата — доброкачественная фолликулярная гиперплазия щитовидной железы (Bethesda — II); тиреоглобулин со смыва пункционной иглы — 0,04 нг/мл. Кальцитонин в крови — в пределах референса. АТ к тиреопероксидазе — отрицательные.
В биохимическом анализе крови — выраженное повышение маркеров миодеструкции креатинфосфокиназа (КФК) — 1467 Ед/л (30–200), лактатдегидрогеназа (ЛДГ) — 324 Ед/л (125–220).
Пациент консультирован неврологом, заподозрена миодистрофия с псевдогипертрофией мышц. Выполнена МРТ головного мозга с в/в контрастированием, по результатам которой визуализировалась аденома гипофиза с инфраселлярным распространением (размерами 17х7,5 мм), единичные очаги в глубоком белом веществе лобных и теменных долей, расширение внутренних ликворных пространств. Объемное образование хиазмально-селлярной области расценено как гиперплазия тиреотрофных клеток аденогипофиза на фоне первичного гипотиреоза. Через 6 месяцев рекомендовано контрольное МРТ-исследование с оценкой динамики размеров опухоли на фоне медикаментозной компенсации тиреоидного статуса.
Учитывая вышеуказанные жалобы, анамнестические данные, результаты физикального и лабораторно-инструментальных обследований, предварительно диагностирован СКДС, включающий в себя миопатию с псевдогипертрофией на фоне длительной декомпенсации первичного гипотиреоза, в отделении инициирована заместительная терапия левотироксином натрия в стартовой дозе 50 мкг/сут, с увеличением до 100 мкг/сут.
Пациент выписался из стационара с улучшением под динамическое наблюдение эндокринолога по месту жительства. Рекомендовано обследование в ФГБНУ «Научный центр неврологии».
Через 2 месяца от начала терапии — медикаментозная компенсация первичного гипотиреоза: ТТГ — 1,134 мМЕ/л (0,4–4,0), свТ4 — 12,95 пмоль/л (7,0–17,6). Уровни маркеров мышечной деструкции все еще оставались повышенными, хотя и имели положительную динамику в виде снижения ЛДГ до 233 Ед/л (135–225), КФК — 737 Ед/л (норма — менее 190). Пациент отмечал улучшение общего самочувствия, регресс симптомов гипотиреоза, однако сохранялись жалобы на боли и слабость мышц нижних конечностей, сложности с самостоятельным передвижением.
Пациенту выполнена МРТ мышц нижних конечностей — МРТ мышц голени: МР-сигнал на Т1 импульсной последовательности не изменен. На Т2 dixon water повышен от задней группы мышц. МРТ мышц бедра: МР-сигнал на Т1 импульсной последовательности изменен во всех mm. Adductors. На Т2 dixon water повышен от mm. Adductors и m. Biceps femoris. Таким образом, МР-картина соответствует проявлениям поясно-конечностной мышечной дистрофии (ПКМД), наиболее вероятные формы — дисферлинопатия. Для подтверждения диагноза и уточнения типа мышечной дистрофии рекомендовано проведение молекулярно-генетического исследования. Пациенту выполнено полное секвенирование экзома методом NGS (массовое параллельное секвенирование). Секвенирование выполнено на платформе Illumina методом парно-концевого чтения (2x100 п.о.). Средняя глубина покрытия — 217,79x, процент целевых нуклеотидов с эффективным покрытием >10х — 99,97%. В результате обнаружены гомозиготные мутации в гене MICU1 — описаны при миопатии с экстрапирамидными симптомами (OMIM: 615673) с аутосомно-рецессивным типом наследования и в гене IGSF1 (NM 001555.5) в 8 экзоне обнаружен ранее не описанный в литературе вариант в гемизиготном состоянии, приводящий к аминокислотной замене p.(Lys420Thr). Гемизиготные мутации в гене IGSF1 описаны при центральном гипотиреозе с увеличением яичек (OMIM: 300888) с Х-сцепленным типом наследования. Однако данный дефект нельзя расценивать как клинически значимый, так как у пациента значительно повышен ТТГ, что свидетельствует о первичном гипотиреозе и исключает центральный характер заболевания, при котором уровень ТТГ снижен или неадекватно нормальный при сниженной функции щитовидной железы.
Через 6 месяцев отмечалась медикаментозная компенсация первичного гипотиреоза: ТТГ — 0, 260 мМЕ/л (0,4–4,0), свТ4 — 13,67 пмоль/л (7,0–17,6). Уровни маркеров мышечной деструкции оставались повышенными, хотя и имели положительную динамику в виде снижения ЛДГ до 280 Ед/л (135–225), КФК — 1643 Ед/л (норма — менее 190).
На фоне терапии восстановление половой функции, тестостерон — 22,47 ммоль/л (6,07–27,1), пролактин — 142,17 мМЕ/л (74,2–339,2). Доза левотироксина натрия уменьшена до 50 мкг/сут.
Патология щитовидной железы является одной из самых часто встречающихся в структуре эндокринных заболеваний [6]., По результатам крупного популяционного исследования, распространенность гипотиреоза составила 4,6%, из которых 0,3% приходится на манифестный (клинически явный), а 4,3% — субклинический [7]. Однако из-за отсутствия специфических симптомов заболевания и сходства проявлений гипотиреоза со многими психическими и соматическими болезнями его диагностика может предоставлять значительные трудности и нередко является несвоевременной, ухудшая качество жизни пациентов. СКДС — одна из разновидностей миопатии, ассоциированная с гипотиреозом, наблюдается в детстве и связана с генерализованной мышечной гипертрофией, микседемой, низкорослостью и кретинизмом [10].
У нашего пациента наблюдалась постепенно прогрессирующая двусторонняя слабость нижних конечностей в сочетании с другими системными проявлениями недостаточности тиреоидных гормонов, такими как общая слабость, восковидный цвет и одутловатость лица, хронические запоры наряду со значительно повышенным уровнем ТТГ, что при поступлении расценено как проявления гипотиреоидной миопатии. Учитывая вышеуказанные жалобы, анамнестические данные, результаты физикального и лабораторно-инструментальных обследований, предварительно диагностирован СКДС, включающий в себя миопатию с псевдогипертрофией на фоне длительной декомпенсации первичного гипотиреоза. Однако сохраняющиеся жалобы на мышечную слабость и повышение уровня маркеров миодеструкции на фоне терапии левотироксином натрия, даже после нормализации ТТГ, привели к поиску других причин разрушения мышечной ткани, не связанных с эндокринопатиями.
Гипотиреоидная миопатия, поражающая 30–80% пациентов с гипотиреозом, часто имитирует полимиозит, что создает трудности в диагностике. Пациентам с мышечной слабостью, миалгией и повышенным уровнем мышечных ферментов, без типичных клинических проявлений гипотиреоза, часто ошибочно ставят диагноз миопатии другой этиологии. Повышенный уровень КФК и ЛДГ еще больше усложняет клиническую картину, часто приводя к ошибочному диагнозу, как полимиозит или другие миопатии [8].
У нашего пациента также отмечались повышенные уровни КФК и ЛДГ, показатели которых уменьшались по мере компенсации гипотиреоза, что является патогномоничным признаком гипотиреоидной миопатии [3–5]. Однако при динамическом наблюдении, несмотря на улучшение общего самочувствия, у пациента сохранялись жалобы на слабость мышц нижних конечностей, а также отмечалось повышение КФК и ЛДГ.
В связи с чем пациенту выполнено полное секвенирование экзома методом NGS, по результатам которого в гене MICU1 (NM 001195518.2) в 9 экзоне обнаружен ранее не описанный в литературе вариант (HG38, chr10:72423313dup, c.992dup) в гомозиготном/гемизиготном состоянии, приводящий к вставке 1 нуклеотида и сдвигу рамки считывания p.(Leu332ThrfsTer44) с глубиной покрытия 200х. Вариант, с большой вероятностью, приводит к потере функции соответствующей копии гена и по совокупности сведений расценивается как вероятно патогенный.
Миопатия с экстрапирамидными симптомами (MPXPS; OMIM #615673) представляет собой аутосомно-рецессивное заболевание, возникающее в результате гомозиготной мутации в гене митохондриального захвата кальция 1 (MICU1), расположенном на хромосоме 10q22, которое вызывает очень редкое нейрональное и мышечное расстройство у людей, характеризующееся нарушением когнитивных функций, ранней мышечной слабостью, повышением уровня креатинкиназы в сыворотке и экстрапирамидными двигательными расстройствами [9, 10].
Регуляция кальция в скелетных мышцах и гомеостаз играют ключевую роль в сопряжении возбуждения и сокращения, расслаблении, восстановлении сарколеммы и адаптации к физической нагрузке. На это влияет доставка Ca 2+ в митохондриальный матрикс через митохондриальный Ca 2+ uniporter (MICU). MICU1 является ключевым регулятором MICU, определяя уровень кальция, и действует как активатор или ингибитор поглощения кальция митохондриями. Мутация потери функции в MICU1 приводит к нервно-мышечному заболеванию с частотой встречаемости менее 1/1000 000 [10–16].
На сегодняшний день в HGMD (базе данных мутаций генов человека) описано 13 патогенных вариантов гена MICU1. Доступно клиническое описание 34 пациентов с 8 различными вариантами гена MICU1 на основе базы данных HGMD. Для остальных 9 пациентов имеется только информация о типе варианта и ограниченная доступная клиническая информация. У большинства пациентов отмечались задержка моторики и речи, затруднения в обучении, повышенный уровень КФК и печеночных трансаминаз, мышечная гипотония, мышечная слабость и прогрессирующие экстрапирамидные симптомы [10][13–17].
Уникальность представленного нами клинического случая заключается в редком сочетании гипотиреоидной миопатии и миопатии с экстрапирамидными симптомами, вызванной мутацией в гене митохондриального захвата кальция 1 (MICU1).
Длительный декомпенсированный гипотиреоз может сопровождаться развитием вторичной гиперплазии тиреотрофов, которая мимикрирует под опухоль гипофиза [18–19]. Учитывая нормализацию уровня пролактина и тестостерона, мы предполагаем, что на фоне лечения у пациента уменьшилось образование гипофиза, которое, вероятно, было не аденомой, а компенсаторной гиперплазией/гипертрофией тиреотрофов на фоне длительно текущего гипотиреоза. У нашего пациента также наблюдался феномен макроТТГ, что может приводить к избыточной терапии левотироксином натрия [20].
Представленное нами клиническое наблюдение вместе с ранее опубликованными данными усиливает доказательства того, что болезни, связанные с патогенными мутациями в гене MICU1, имеют определенную клиническую картину. Идентификация большего числа таких генетических вариантов и более полное описание симптомов заболевания ускоряют процесс молекулярной диагностики. Это, в свою очередь, улучшает качество генетического консультирования для пар, входящих в группу риска, и предоставляет им возможность минимизировать вероятность рождения детей с этим заболеванием.
Источники финансирования. Статья подготовлена в рамках тематики государственного задания на выполнение НИР (№12302300097-0).
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи.
Участие авторов. Дзеранова Л.К. — получение, анализ данных, внесение в рукопись важной правки; Колодкина А.А. — внесение в рукопись важной правки, одобрение финальной версии; Пигарова Е.А. — получение, анализ данных, внесение в рукопись важной правки; Шутова А.С. — получение, анализ данных, написание статьи; Перепелова М.А. — получение, анализ данных, написание статьи; Слоква В.К. — получение, анализ данных; внесение в рукопись важной правки; Перепелов А.В. — генетическое консультирование, внесение в рукопись важной правки.
Все авторы одобрили финальную версию статьи перед публикацией, выразили согласие нести ответственность за все аспекты работы, подразумевающую надлежащее изучение и решение вопросов, связанных с точностью или добросовестностью любой части работы.
Согласие пациента. Пациент добровольно подписал информированное согласие на публикацию персональной медицинской информации в обезличенной форме в журнале «Ожирение и метаболизм».
1. Ильченко В.А., Лебедева А.О., Гордиенко Б.В., Болотин Е.В. «Маски» гипотиреоза (описание клинического случая) // Альманах клинической медицины. — 2014. — №35.
2. Муравьева Г.В., Девликамова Ф.И. Нервно-мышечные осложнения при заболеваниях щитовидной железы. // Практическая медицина. — 2013. — №1. — С. 38-41.
3. Lockshin MD. Endocrine origins of rheumatic disease. Diagnostic clues to interrelated syndromes. Postgrad Med. 2002;111(4):87-8, 91-2
4. Ruff RL, Weissmann J. Endocrine Myopathies. Neurol Clin. 1988;6(3):575-592. doi: https://doi.org/10.1016/S0733-8619(18)30862-4
5. Thyroid myopathy. Effect of treatment with thyroid hormones / A. Del Palacio [et al.] An Med Interna. 1990;7(3):120-2
6. Фадеев В.В. Гипотиреоз / В.В. Фадеев, Г.А. Мельниченко. — М., РКИ Северо-пресс, 2004. — 286 с.
7. Hollowell JG, Staehling NW, Flanders WD, Hannon WH, Gunter EW, Spencer CA, Braverman LE. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab. 2002 Feb;87(2):489-99. doi: https://doi.org/10.1210/jcem.87.2.8182.
8. Qiang F, He Q, Wang L, Sheng J. Polymyositis-like hypothyroid myopathy: diagnostic challenges and therapeutic outcomes in a case series. Clin Exp Med. 2025;25(1):286. doi: https://doi.org/10.1007/s10238-025-01828-3
9. Logan CV, Szabadkai G, Sharpe JA, Parry DA, Torelli S, Childs AM, et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat Genet. 2014;46(2):188–193 doi: https://doi.org/10.1038/ng.2851
10. Debattisti V, Horn A, Singh R, Seifert EL, Hogarth MW, Mazala DA et al (2019) Dysregulation of mitochondrial Ca2+ uptake and sarcolemma repair underlie muscle weakness and wasting in patients and mice lacking MICU1. Cell Rep 29(5):1274–1286.e6. https://doi.org/10.1016/j.celrep.2019.09.063
11. Tufi R, Gleeson TP, von Stockum S, Hewitt VL, Lee JJ, Terriente-Felix A, et al. Comprehensive genetic characterization of mitochondrial Ca 2+ uniporter components reveals their different physiological requirements in vivo. Cell Rep 2019;27(5):1541–1550.e5. doi: https://doi.org/10.1016/j.celrep.2019.04.033
12. Llorente-Folch I, Rueda CB, Pardo B, Szabadkai G, Duchen MR, Satrustegui J. The regulation of neuronal mitochondrial metabolism by calcium. J Physiol. 2015;593(16):3447–3462. doi: https://doi.org/10.1113/JP270254
13. Burgoyne RD, Haynes LP. Understanding the physiological roles of the neuronal calcium sensor proteins. Mol Brain. 2012;5(1):1–11. doi: https://doi.org/10.1186/1756-6606-5-2
14. Kawamoto EM, Vivar C, Camandola S. Physiology and pathology of calcium signaling in the brain. Front Pharmacol. 2012;3 APR(April):1–17. doi: https://doi.org/10.3389/fphar.2012.00061
15. Ryglewski S, Pflueger HJ, Duch C. Expanding the neuron’s calcium signaling repertoire: Intracellular calcium release via voltageinduced PLC and IP3R activation. PLoS Biol. 2007;5(4):818–827. doi: https://doi.org/10.1371/journal.pbio.0050066
16. Arvizo RR, Moyano DF, Saha S, Thompson MA, Bhattacharya R, et al. Probing novel roles of the mitochondrial uniporter in ovarian cancer cells using nanoparticles. J Biol Chem. 2013;288(24):17610–17618. doi: https://doi.org/10.1074/jbc.M112.435206
17. Chaudhuri D, Sancak Y, Mootha VK, Clapham DE. MCU encodes the pore conducting mitochondrial calcium currents. Elife. 2013(2):4–11. doi: https://doi.org/10.7554/eLife.00704
18. Young M, Kattner K, Gupta K. Pituitary hyperplasia resulting from primary hypothyroidism mimicking macroadenomas. Br J Neurosurg. 1999;13(2):138-42. doi: https://doi.org/10.1080/02688699943880
19. Horvath E, Kovacs K, Scheithauer BW. Pituitary hyperplasia. Pituitary. 1999;1(3-4):169-79. doi: https://doi.org/10.1023/a:1009952930425
20. Сазонова Д.В., Перепелова М.А., Шутова А.С., Колесникова Г.С., Никанкина Л.В., Пигарова Е.А., Дзеранова Л.К. Сочетание феноменов макро-ТТГ и макропролактинемии у пациента с аутоиммунным тиреоидитом и витилиго // Проблемы эндокринологии. — 2024. — Т. 70. — №5. — С. 34-39. doi: https://doi.org/10.14341/probl13390
Перепелова Маргарита Александровна, врач-эндокринолог
117292, Москва, ул. Дм. Ульянова, д. 11
Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи
Слоква Валерия Константиновна, врач-эндокринолог
г. Москва
Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи
Пигарова Екатерина Александровна, врач-эндокринолог, д.м.н.
г. Москва
Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи
Шутова Александра Сергеевна, врач-эндокринолог, к.м.н.
г. Москва
Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи
Колодкина Анна Александровна, врач-эндокринолог, к.м.н.
г. Москва
Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи
Перепелов Александр Васильевич, врач-генетик, к.м.н.
г. Обнинск
Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи
Дзеранова Лариса Константиновна, врач-эндокринолог, д.м.н.
г. Москва
Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи
|
|
1. Рисунок 1. Пациент Э., 26 лет. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(1MB)
|
Метаданные ▾ | |
Перепелова М.А., Слоква В.К., Пигарова Е.А., Шутова А.С., Колодкина А.А., Перепелов А.В., Дзеранова Л.К. Гипотиреоидная миопатия и ее связь с мутациями в гене MICU1: клинический случай. Ожирение и метаболизм. 2025;22(3):263-268. https://doi.org/10.14341/omet13287
Perepelova M.A., Slokva V.K., Pigarova E.A., Shutova A.S., Kolodkina A.A., Perepelov A.V., Dzeranova L.K. Hypothyroid myopathy and its association with MICU1 gene mutations: a clinical caseauthors. Obesity and metabolism. 2025;22(3):263-268. (In Russ.) https://doi.org/10.14341/omet13287
 |
117292
Россия, Москва, ул. Дм. Ульянова, д.11
____________________________________
117292
11, Dm. Ul’yanova str., Moscow, Russian Federation



































