




Содержание
Перейти к:
https://doi.org/10.14341/omet12750
Перейти к:
В данной обзорной статье представлены сведения из литературных источников, которые дают представление о связи метаболических нарушений, возникающих на фоне ожирения, с эндотоксинемией, а также влиянии этих состояний на поддержание низкоинтенсивного воспаления в организме. Приведено описание гормональной и иммунной перестройки белой жировой ткани, основных путей поступления и метаболизма эндотоксина. Особое внимание уделено механизмам взаимного влияния ожирения и эндотоксинемии. Описанные М.Ю. Яковлевым в 1988 г. «эндотоксиновая агрессия» и P.D. Cani и соавт. в 2007 г. «метаболическая эндотоксинемия», на наш взгляд, являются одними из важнейших триггеров развития и прогрессирования целого спектра острых и хронических заболеваний. Исходя из данных последних лет, жировая ткань представляет собой активный эндокринный орган, способный оказывать влияние как на обменные процессы, так и на состояние врожденных и приобретенных механизмов иммунной защиты. В настоящее время доказано, что высококалорийные диеты приводят к увеличению не только избыточной массы тела, но и уровня циркулирующего в крови эндотоксина. Углубленное изучение способности ожирения и эндотоксинемии потенцировать взаимное провоспалительное действие может помочь как в понимании патогенеза основных сердечно-сосудистых, аутоиммунных, аллергических и инфекционных (в том числе вирусных) заболеваний, так и в разработке методов нефармакологической и медикаментозной коррекции данных состояний.
Белоглазов В.А., Яцков И.А., Кумельский Е.Д., Половинкина В.В. Метаболическая эндотоксинемия: возможные причины и последствия. Ожирение и метаболизм. 2021;18(3):320-326. https://doi.org/10.14341/omet12750
Beloglazov V.A., Yatskov I.A., Kumelsky E.D., Polovinkina V.V. Metabolic endotoxemia: possible causes and consequences. Obesity and metabolism. 2021;18(3):320-326. (In Russ.) https://doi.org/10.14341/omet12750
Ожирение является серьезным фактором, отягощающим многие острые и хронические патологические процессы, происходящие в организме. Приблизительно 65% взрослого населения США и более 100 млн человек во всем мире имеют избыточный вес или ожирение [1][2]. Данные последних лет свидетельствуют о том, что жировая ткань является не только депо энергетического материала, но и достаточно сложно устроенным иммунным и эндокринным органом [3]. Ожирение характеризуется низкоинтенсивным воспалением, связанным с повышением уровня системных и местных провоспалительных цитокинов [4]. Подтверждена роль иммунного дисбаланса при ожирении в развитии диабета II типа (СД2), метаболического синдрома и сердечно-сосудистых заболеваний [5][6]. Также интерес представляет взаимосвязь параметров метаболических нарушений и эндотоксинемии, что, по данным отечественных и зарубежных исследований, может потенцировать многочисленные нарушения эффекторных механизмов поддержания гомеостаза [7–10].
Путем анализа баз данных MEDLINE (PubMed) по данным на май 2021 г. был проведен поиск по ключевым словам «metabolic endotoxemia», «endotoxin and obesity» и «LPS and obesity», а также поиск в библиотеке eLibrary по ключевым словам «метаболическая эндотоксинемия» и «ожирение и эндотоксинемия». Большинство научных статей, представленных в данном литературном обзоре, опубликовано за последние 5 лет.
Для понимания роли жировой ткани в регуляции иммунных процессов необходимо знать основные изменения, происходящие в белой жировой ткани (БЖТ) у худых людей и лиц с ожирением. БЖТ депонируется под кожей, в брюшной полости и в парависцеральной клетчатке большинства органов. У мужчин 10–20% жировой ткани расположено висцерально, тогда как у женщин — только 5–8% [11]. Между подкожной и висцеральной БЖТ существует множество физиологических различий; адипоциты висцеральной БЖТ более инсулинорезистентны, метаболически активны и обладают большей липолитической активностью [3]. Кроме того, накопление висцерального жира связано с повышенным риском развития сахарного диабета 2 типа (СД2) и метаболического синдрома [12–14]. Хотя БЖТ в основном состоит из адипоцитов, она также содержит преадипоциты, иммунные клетки, фибробласты и сосудистые клетки. Количество и фенотип этих клеток изменяются в зависимости от локализации жировой ткани, а также различаются у людей с ожирением и худых [15]. БЖТ у худых обычно состоит из иммунных клеток, которые являются преимущественно регуляторными и иммуносупрессивными по своей природе, включая M2-подобные макрофаги жировой ткани (ATM), регуляторные T-клетки (Treg), T-хелперы (Th) типа 2, инвариантные натуральные киллеры (iNKT) и эозинофилы. У худых людей ATM являются преобладающими иммунными клетками, присутствующими в БЖТ, и составляют 5–15% от общего количества клеток [16]. ATM M2 равномерно распределены в жировой ткани и выполняют различные физиологические функции, в том числе способствуют удалению мертвых адипоцитов, ингибируют пролиферацию предшественников адипоцитов и секретируют противовоспалительные цитокины, такие как интерлейкины (IL-10, IL-4, IL-13 и IL-1Rα) [17][18].
При ожирении происходит повышенное накопление липидов, что приводит к гипертрофии адипоцитов, гипоксии и повышенной гибели клеток. Дисфункция жировой ткани способствует изменению микроокружения, в котором увеличивается секреция провоспалительных цитокинов, включая фактор некроза опухоли альфа (TNF-α), интерлейкин-6 (IL-6), интерлейкин-8 (IL-8) и моноцитарный хемоаттрактантный протеин-1 (MCP-1), и других хемокинов, продуцируемых адипоцитами и иммунными клетками, что способствует усиленной миграции циркулирующих моноцитов и других клеток врожденного и адаптивного иммунитета в жировую ткань [19–21]. Повышенная инфильтрация моноцитами [16] и задержка в тканях макрофагов [22] способствуют значительному увеличению количества клеток моноцитарно-макрофагального ряда у пациентов с ожирением. Помимо увеличения количества макрофагов, провоспалительная среда в БЖТ у пациентов с ожирением способствует изменениям фенотипов ATM [18]. Первоначально считалось, что ожирение приводит к увеличению концентрации M1-провоспалительных макрофагов в БЖТ. Но недавно было обнаружено, что в БЖТ у пациентов с ожирением, кроме M1 макрофагов, присутствуют уникальные «метаболически активные макрофаги» с отчетливым провоспалительным профилем [23]. При ожирении в БЖТ увеличивается содержание тучных и дендритных клеток (DC), CD4+ Th1- и Th17-клеток и CD8+ цитотоксических Т-лимфоцитов [24–28]. Изменение баланса провоспалительных и противовоспалительных адипокинов и цитокинов представлено на рисунке 1.
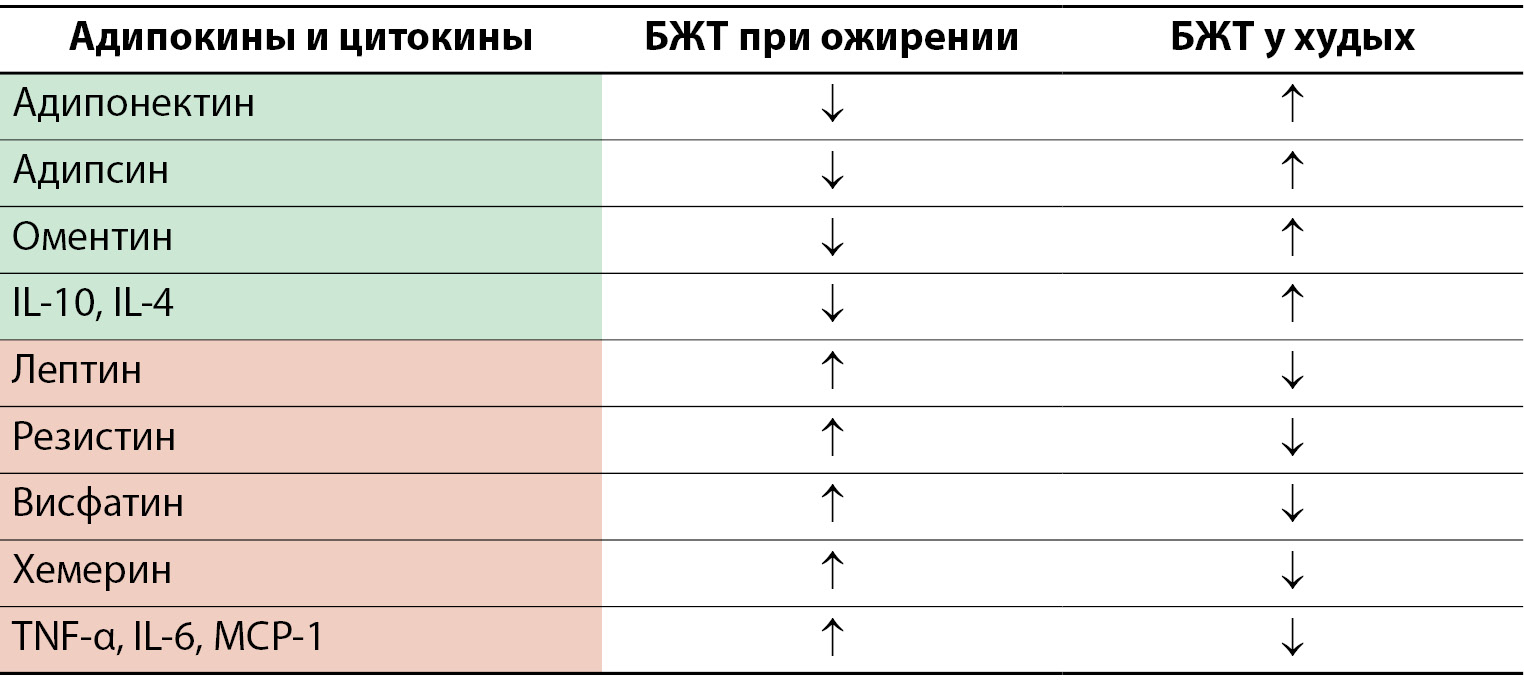
Рисунок 1. Изменение баланса провоспалительных и противовоспалительных адипокинов и цитокинов при ожирении.
Клинические данные свидетельствуют о том, что частота аутоиммунных заболеваний увеличивается параллельно с интенсивностью ожирения и метаболического синдрома [29]. В поддержку этой концепции, ожирение, вызванное диетой, усугубляет проявления аутоиммунных заболеваний, что было убедительно продемонстрировано на экспериментальных моделях животных, включая воспалительные заболевания кишечника [30], коллаген-индуцированный артрит [31], экспериментальный аутоиммунный энцефаломиелит (EAE, модель рассеянного склероза) [32, 33] и системную красную волчанку (СКВ) [34].
Эндотоксины представляют собой термостойкие липополисахариды (LPS), которые являются основным гликолипидным компонентом внешней мембраны грамотрицательных бактерий [35], составляющих приблизительно 70% от общего количества бактерий в кишечнике [36].
Взаимодействие моноцитарно-макрофагальных клеток с LPS в основном осуществляется через рецепторы mCD14 и TLR-4/MD-2 (толл-подобные рецепторы 4 типа с адаптерным белком — MD-2).
К гуморальным LPS-связывающим системам относятся: LPS-связывающий белок (LBP), анти-LPS-антитела, растворимые CD14 (sCD14) рецепторы, С-реактивный белок (CRP), амилоид А, лизоцим, липопротеины высокой плотности (ЛПВП), липопротеины низкой плотности (LDL), белок теплового шока HSP 60, интерферон, альбумин, лактоферрин, аполипопротеины (ApoB, ApoA-I, ApoE), фибронектин, гликопротеины (САР18, САР33), антитела к Re-гликолипиду (глубокая детерминанта R-кора), состоящие из липида А и кетодезоксиоктанта [37].
Липид A является наиболее важной частью LPS и основным центром иммуностимулирующей способности LPS, поскольку он специфически распознается комплексом TLR4/MD-2 [38][39]. Наибольшей способностью к активации комплекса TLR4/MD-2 и последующему запуску провоспалительного ответа обладает гексаацилированный липид A [39–41]. Провоспалительная активность коррелирует со способом связывания липида A с комплексом TLR4/MD-2 [42]. В настоящее время имеются лишь ограниченные данные о нескольких формах LPS с антивоспалительным эффектом, способных конкурировать с токсичным LPS за связывание с TLR4/MD-2, предотвращая, таким образом, генерацию провоспалительного ответа [41]. К таким формам относится тетраацильный липид А, который свойствен определенным видам бактерий рода Bacteroides [41][43][44].
LPS могут перемещаться в систему кровообращения посредством прямой диффузии из-за кишечной парацеллюлярной проницаемости или в составе хиломикрон [45]. В дальнейшем LPS может транспортироваться в гепатоциты, связываясь с ЛПВП, ЛПНП или хиломикронами для последующей инактивации (деацилирования) и экскреции с желчью [45][46] (рис. 2). Ряд состояний, таких как стресс, вирусные заболевания, дисбактериоз кишечника, погрешности в диете и антибиотикотерапия, могут увеличивать объем поступающего в кровоток LPS — индуцировать системное воспаление, что было постулировано М.Ю. Яковлевым еще в конце XX в. [47]. Согласно исследованию d’Hennezel и соавт., у лиц с ожирением дисбаланс кишечной микрофлоры приводит к увеличению поступления в системный кровоток LPS с гексаацильным липидом А [48]. Кроме этого, известно, что диета с высоким содержанием жиров увеличивает проницаемость кишечника с помощью различных механизмов: изменяет распределение и снижает экспрессию плотных контактов, индуцирует апоптоз эпителиальных клеток кишечника, прямо и косвенно стимулирует провоспалительные сигнальные каскады, увеличивая продукцию разрушающих эпителиальный слой цитокинов, уменьшая уровень барьерных цитокинов; отрицательно модулирует состав кишечной слизи и обогащает микрофлору кишечника видами, разрушающими слизистый барьер [49][50].
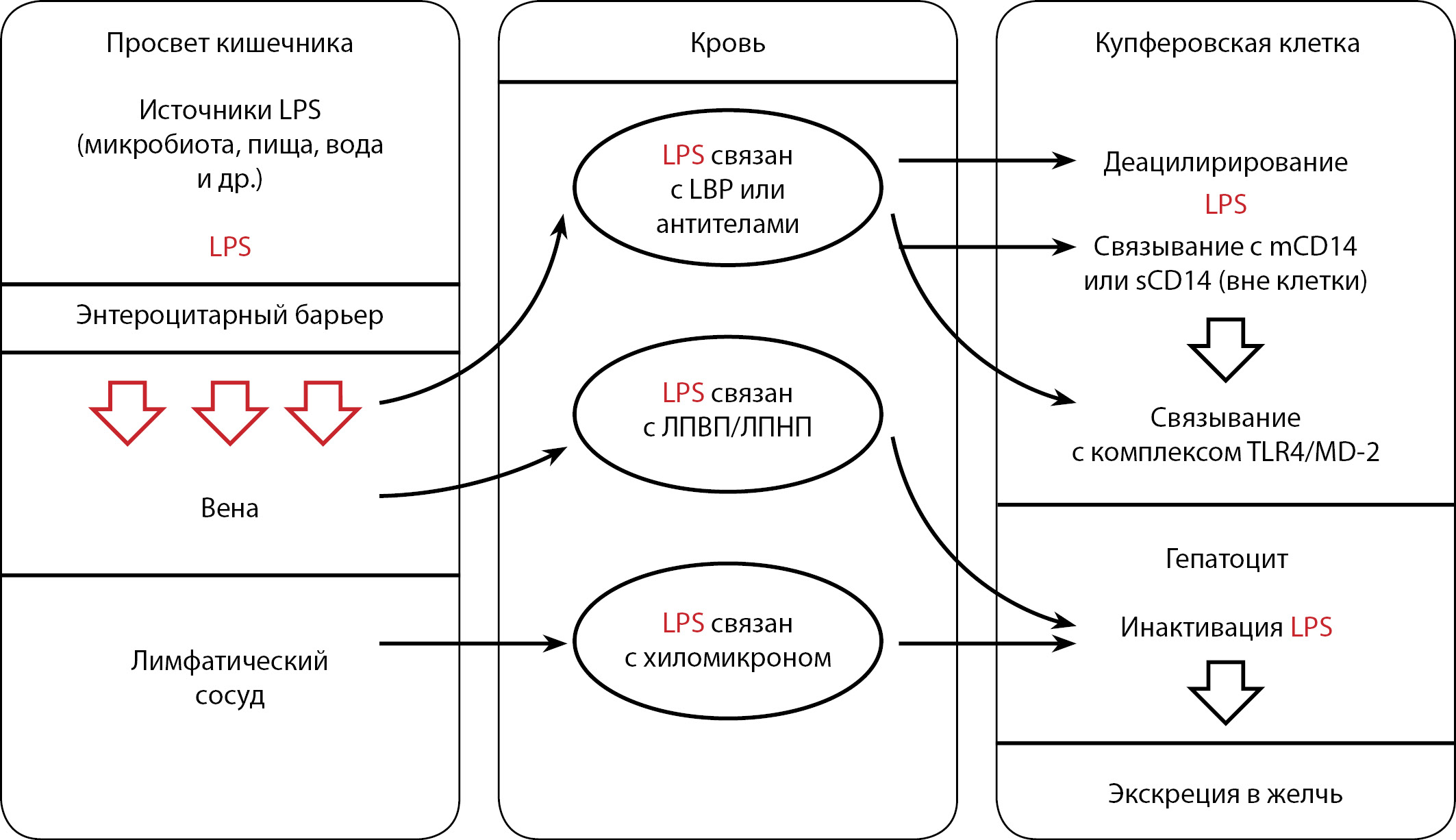
Рисунок 2. Основные пути поступления и метаболизма LPS.
LPS — липополисахарид; LBP — липополисахарид-связывающий белок; ЛПВП — липопротеины высокой плотности; ЛПНП — липопротеины низкой плотности; mCD14 — сцепленный с мембраной CD14; sCD14 — растворимая форма CD14; TLR4/MD-2 — рецепторный комплекс, распознающий липополисахарид.
Cani и соавт. описали так называемую «метаболическую эндотоксинемию» как состояние хронически повышенного уровня LPS в плазме крови на уровне в 10–50 раз ниже, чем при септических состояниях [51]. Метаболическая эндотоксинемия как наблюдалась у генетически страдающих ожирением мышей, потребляющих нормальный корм, так и индуцировалась у тощих мышей, потребляющих высококалорийную диету [51]. Повышение уровня эндотоксина, вызванное высококалорийной диетой, было связано с повышенным отложением жира, повышением активации провоспалительных каскадов, перекисного окисления и выработки резистентности к инсулину [11][51]. Эти результаты доказывают, что кишечный эндотоксин является важнейшим индуктором повышенной провоспалительной активности в экспериментальной модели ожирения у грызунов [11].
Роль эндотоксина как медиатора развития жировой ткани, системных и локальных воспалительных процессов и метаболических нарушений была подтверждена в эксперименте при введении низких доз LPS худым мышам, которые находились на стандартной диете [51]. Инъекция 300 мкг/кг/день LPS вызывала у худых мышей сходные нарушения, как и при ожирении, вызванном высококалорийной диетой. Кроме того, худые мыши, лишенные кластера дифференцировки CD14, были устойчивы к увеличению веса, вызванному диетой с высоким содержанием жиров, тканеспецифическому воспалению, отложению липидов в печени и резистентности к инсулину [51]. LPS может инициировать системное и локальное воспаление, а также приводить к продукции активных форм кислорода (ROS) при связывании с TLR4 и последующей активации NF-κB [52–54]. TLR4 широко экспрессируется на иммунных клетках, клетках печени, жировой ткани и в скелетных мышцах [55–57]. В совокупности эти ткани играют важную роль в регуляции гомеостаза глюкозы и липидов. Установлено, что провоспалительные цитокины и продуцирование ROS влияют на нормальный метаболизм в этих тканях [27–30]. Так, Cani и соавт. сообщили о повышенной выработке провоспалительных цитокинов (например, TNFa, IL-6, IL-1), усилении окислительного стресса (НАДФH-оксидаза, индуцируемая синтаза оксида азота) и увеличении маркеров инфильтрации макрофагов (CD86) в ткани печени у мышей с ожирением и метаболической эндотоксинемией [25].
В то время как существует большое количество зарубежных и отечественных работ по изучению эндотоксинемии на животных моделях, данных о взаимосвязи LPS и метаболических заболеваний у людей на данный момент значительно меньше.
LPS присутствует в низких концентрациях и у здоровых людей, но даже однократный прием пищи с высоким содержанием жиров уже может значительно увеличить уровни циркулирующего в крови эндотоксина [58][59]. В исследованиях последних лет сообщается о повышенных уровнях LPS и LBP у пациентов с метаболическим синдромом или СД2 [7][60–62]. Так, Pussinen и соавт. проанализировали уровни LPS у пациентов с СД2 и сравнили их с контрольной группой, в которой около 20% пациентов имели метаболический синдром. Уровень эндотоксина был значительно повышен улиц с СД2 [7]. Парентеральное введение LPS провоцировало развитие инсулинорезистентности и системного воспаления [8], а интервенция с перееданием продолжительностью 8 нед была связана с повышением уровня эндотоксина в плазме крови, что подтверждает связь избыточного питания с эндотоксинемией и развитием инсулинорезистентности [9]. В недавнем исследовании Cox и соавт. использовали LPS, LBP, а также белок, связывающий жирные кислоты кишечника (iFABP), для расчета показателя риска кишечной проницаемости, который был повышен улиц с СД2 [63]. Также наличие у пациентов неалкогольной жировой болезни печени (НАЖБП) было связано с еще более высоким уровнем эндотоксина в крови [64].
В нашем предшествующем исследовании у пациентов, коморбидных по сезонному аллергическому риниту (САР), артериальной гипертензии (АГ) и ожирению выявлено повышение в периферической крови уровня рецепторов СРБ, LBP и sCD14, что свидетельствует о наличии системного хронического воспаления и участии провоспалительных LPS-связывающих механизмов в персистенции данного воспаления. Также было установлено наличие связи между концентрацией СРБ и LBP у пациентов с ожирением в период поллинации причинно-значимых аллергенов, что свидетельствует о наличии функциональной взаимосвязи обострения локального T2-аллергического воспаления, системного воспаления и провоспалительного ответа ЛПС-связывающих систем при метаболической эндотоксинемии [65].
Высокий уровень циркулирующего эндотоксина вследствие увеличения кишечной проницаемости и нарушения микробиома, а также дисбаланс LPS-связывающих систем являются клинически значимыми факторами в развитии сердечно-метаболических заболеваний. Например, повышенная базовая концентрация эндотоксина в сыворотке натощак явилась предиктором кардиоваскулярных событий в течение 10 лет у лиц с ишемической болезнью сердца [65]. Кроме того, риск развития диабета увеличился у здоровых лиц в возрасте 25–75 лет с повышенным уровнем эндотоксина в сыворотке крови при последующем динамическом наблюдении на протяжении 10 лет [7]. В китайском когортном исследовании было доказано, что риск развития метаболического синдрома и большинства его компонентов (включая ожирение) повышен у людей среднего возраста и пожилых с повышенным уровнем LBP [66].
В связи с увеличением доли населения, страдающего избыточным весом и ожирением, проблема гормональной и иммунной перестройки жировой ткани, а также влияния высококалорийных диет и ожирения на уровень эндотоксина в крови становится все более актуальной. Данные метаболические нарушения могут стать триггерами развития и прогрессирования целого спектра острых и хронических заболеваний, приводить в последующем к увеличению инвалидизации и смертности трудоспособного населения страны. Углубленное изучение способности ожирения и эндотоксинемии потенцировать взаимное провоспалительное действие может помочь как в понимании патогенеза основных сердечно-сосудистых, аутоиммунных, аллергических и инфекционных (в том числе вирусных) заболеваний, так и в разработке методов нефармакологической и медикаментозной коррекции данных состояний.
Источники финансирования. Работа выполнена по инициативе авторов без привлечения финансирования.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи.
Участие авторов. Белоглазов В.А. — концепция статьи, анализ публикаций, интерпретация результатов, внесение существенных правок с целью повышения научной ценности статьи; Яцков И.А. — анализ публикаций, интерпретация результатов, внесение существенных правок с целью повышения научной ценности статьи; Кумельский Е.Д. — сбор и систематизация данных, написание статьи; Половинкина В.В. — сбор и систематизация данных, написание статьи. Все авторы одобрили финальную версию статьи перед публикацией, выразили согласие нести ответственность за все аспекты работы, подразумевающую надлежащее изучение и решение вопросов, связанных с точностью или добросовестностью любой части работы.
1. Flegal KM, Kruszon-Moran D, Carroll MD, et al. Trends in Obesity Among Adults in the United States, 2005 to 2014. JAMA. 2016;315(21):2284-2291. doi: https://doi.org/10.1001/jama.2016.6458
2. Afshin A, Forouzanfar MH, Reitsma MB, et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N Engl J Med. 2017;377(1):13-27. doi: https://doi.org/10.1056/NEJMoa1614362
3. Arner P. Differences in lipolysis between human subcutaneous and omental adipose tissues. Ann Med. 1995;27(4):435-438. doi: https://doi.org/10.3109/07853899709002451
4. Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415-445. doi: https://doi.org/10.1146/annurev-immunol-031210-101322
5. Dandona P, Aljada A, Bandyopadhyay A. Inflammation: The link between insulin resistance, obesity and diabetes. Trends in Immunology. 2004;25:4-7. doi: https://doi.org/10.1016/j.it.2003.10.013
6. Van Gaal LF, Mertens IL, Christophe E. Mechanisms linking obesity with cardiovascular disease. Nature. 2006; 444:875-880. doi: https://doi.org/10.1038/nature05487
7. Pussinen PJ, Havulinna AS, Lehto M, et al. Endotoxemia is associated with an increased risk of incident diabetes. Diabetes Care. 2011;34:392-397. doi: https://doi.org/10.2337/dc10-1676
8. Agwunobi AO, Reid C, Maycock P, et al. Insulin resistance and substrate utilization in human endotoxemia. The Journal of Clinical Endocrinology and Metabolism. 2000;85:3770-3778. doi: https://doi.org/10.1210/jcem.85.10.6914
9. Krogh-Madsen R, Plomgaard P, Akerstrom T, et al. Effect of short-term intralipid infusion on the immuneresponse during low-dose endotoxemia in humans. The Journal of Clinical Endocrinology and Metabolism. 2008;294:371-379. doi: https://doi.org/10.1152/ajpendo.00507.2007
10. Яковлев М.Ю. Системная эндотоксинемия. Гомеостаз и общая патология. — М.: Наука; 2021.
11. Mongraw‐Chaffin M, Hairston KG, Hanley AJG, et al. Association of Visceral Adipose Tissue and Insulin Resistance with Incident Metabolic Syndrome Independent of Obesity Status: The IRAS Family Study. Obesity. 2021;29(7):1195-1202. doi: https://doi.org/10.1002/oby.23177
12. Kissebah AH, Vydelingum N, Murray R, et al. Relation of body fat distribution to metabolic complications of obesity. J Clin Endocrinol Metab. 1982;54(2):254-260. doi: https://doi.org/10.1210/jcem-54-2-254
13. Lemieux I, Després J-P. Metabolic Syndrome: Past, Present and Future. Nutrients. 2020;12(11):3501. doi: https://doi.org/10.3390/nu12113501
14. Gastaldelli A, Miyazaki Y, Pettiti M, et al. Metabolic effects of visceral fat accumulation in type 2 diabetes. J Clin Endocrinol Metab. 2002;87(11):5098-5103. doi: https://doi.org/10.1210/jc.2002-020696
15. Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11(2):85-97. doi: https://doi.org/10.1038/nri2921
16. Kunz HE, Hart CR, Gries KJ, et al. Adipose tissue macrophage populations and inflammation are associated with systemic inflammation and insulin resistance in obesity. Am J Physiol Metab. 2021;321(1):E105-E121. doi: https://doi.org/10.1152/ajpendo.00070.2021
17. Nawaz A, Aminuddin A, Kado T, et al. CD206+ M2-like macrophages regulate systemic glucose metabolism by inhibiting proliferation of adipocyte progenitors. Nat Commun. 2017;8(1):286. doi: https://doi.org/10.1038/s41467-017-00231-1
18. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175-184. doi: https://doi.org/10.1172/JCI29881
19. Nara N, Nakayama Y, Okamoto S, et al. Disruption of CXC motif chemokine ligand-14 in mice ameliorates obesity-induced insulin resistance. J Biol Chem. 2007;282(42):30794-30803. doi: https://doi.org/10.1074/jbc.M700412200
20. Kochumon S, Al Madhoun A, Al-Rashed F, et al. Elevated adipose tissue associated IL-2 expression in obesity correlates with metabolic inflammation and insulin resistance. Sci Rep. 2020;10(1):16364. doi: https://doi.org/10.1038/s41598-020-73347-y
21. Duffaut C, Zakaroff-Girard A, Bourlier V, et al. Interplay between human adipocytes and T lymphocytes in obesity: CCL20 as an adipochemokine and T lymphocytes as lipogenic modulators. Arterioscler Thromb Vasc Biol. 2009;29(10):1608-1614. doi: https://doi.org/10.1161/ATVBAHA.109.192583
22. Ramkhelawon B, Hennessy EJ, Ménager M, et al. Netrin-1 promotes adipose tissue macrophage retention and insulin resistance in obesity. Nat Med. 2014;20(4):377-384. doi: https://doi.org/10.1038/nm.3467
23. Kratz M, Coats BR, Hisert KB, et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014;20(4):614-625. doi: https://doi.org/10.1016/j.cmet.2014.08.010
24. Bertola A, Ciucci T, Rousseau D, et al. Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes. 2012;61(9):2238-2247. doi: https://doi.org/10.2337/db11-1274
25. Stefanovic-Racic M, Yang X, Turner MS, et al. Dendritic cells promote macrophage infiltration and comprise a substantial proportion of obesity-associated increases in CD11c+ cells in adipose tissue and liver. Diabetes. 2012;61(9):2330-2339. doi: https://doi.org/10.2337/db11-1523
26. Liu J, Divoux A, Sun J, et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med. 2009;15(8):940-945. doi: https://doi.org/10.1038/nm.1994
27. Nishimura S, Manabe I, Nagasaki M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15(8):914-920. doi: https://doi.org/10.1038/nm.1964
28. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87-91. doi: https://doi.org/10.1126/science.7678183
29. Manzel A, Muller DN, Hafler DA, et al. Role of «Western diet» in inflammatory autoimmune diseases. Curr Allergy Asthma Rep. 2014;14(1):404. doi: https://doi.org/10.1007/s11882-013-0404-6
30. Paik J, Fierce Y, Treuting PM, et al. High-fat diet-induced obesity exacerbates inflammatory bowel disease in genetically susceptible Mdr1a-/- male mice. J Nutr. 2013;143(8):1240-1247. doi: https://doi.org/10.3945/jn.113.174615
31. Chehimi M, Vidal H, Eljaafari A. Pathogenic Role of IL-17-Producing Immune Cells in Obesity, and Related Inflammatory Diseases. J Clin Med. 2017;6(7):68. doi: https://doi.org/10.3390/jcm6070068
32. Winer S, Paltser G, Chan Y, et al. Obesity predisposes to Th17 bias. Eur J Immunol. 2009;39(9):2629-2635. doi: https://doi.org/10.1002/eji.200838893
33. Timmermans S, Bogie JF, Vanmierlo T, et al. High fat diet exacerbates neuroinflammation in an animal model of multiple sclerosis by activation of the Renin Angiotensin system. J Neuroimmune Pharmacol. 2014;9(2):209-217. doi: https://doi.org/10.1007/s11481-013-9502-4
34. Hanna Kazazian N, Wang Y, Roussel-Queval A, et al. Lupus Autoimmunity and Metabolic Parameters Are Exacerbated Upon High Fat Diet-Induced Obesity Due to TLR7 Signaling. Front Immunol. 2019;10:2015. doi: https://doi.org/10.3389/fimmu.2019.02015
35. Adamik B, Smiechowicz J, Kübler A. The importance of early detection of endotoxemia. Innate Immunity. 2016;22(7):503-509. doi: https://doi.org/10.1177/1753425916660177
36. Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annual Review of Biochemistry. 2002;71:635-700. doi: https://doi.org/10.1146/annurev.biochem.71.110601.135414
37. Gordienko AI, Beloglazov VA, Kubyshkin AV, et al. Humoral Anti-Endotoxin Immunity Imbalance as a Probable Factor in the Pathogenesis of Autoimmune Diseases. Hum Physiol. 2019;45:337-341. doi: https://doi.org/10.1134/S036211971903006X
38. Di Lorenzo F, Kubik Ł, Oblak A, et al. Activation of Human Toll-like Receptor 4 (TLR4) Myeloid Differentiation Factor 2 (MD-2) by Hypoacylated Lipopolysaccharide from a Clinical Isolate of Burkholderiacenocepacia. Journal of Biological Chemistry. 2015;290(35):21305-21319. doi: https://doi.org/10.1074/jbc.m115.649087
39. Molinaro A, Holst O, Di Lorenzo F, et al. Chemistry of Lipid A: At the Heart of Innate Immunity. Chem - A Eur J. 2015;21(2):500-519. doi: https://doi.org/10.1002/chem.201403923
40. Munford RS. Sensing Gram-Negative Bacterial Lipopolysaccharides: a Human Disease Determinant? Infection and Immunity. 2008;76:454-465. doi: https://doi.org/10.1128/iai.00939-07
41. Di Lorenzo F, Palmigiano A, Al Bitar-Nehme S, et al. The Lipid A from Rhodopseudomonaspalustris Strain BisA53 LPS Possesses a Unique Structure and Low Immunostimulant Properties. Chemistry. 2017;23(15):3637-3647. doi: https://doi.org/10.1002/chem.201604379
42. Park BS, Song DH, Kim HM, et al. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191-1195. doi: https://doi.org/10.1038/nature07830
43. Lembo-Fazio L, Billod JM, Di Lorenzo F, et al. Bradyrhizobium Lipid A: Immunological Properties and Molecular Basis of Its Binding to the Myeloid Differentiation Protein-2/Toll-Like Receptor 4 Complex. Frontiers in Immunology. 2018;9:1888. doi: https://doi.org/10.3389/fimmu.2018.01888
44. Pallach M, Di Lorenzo F, Facchini FA, et al. Structure and inflammatory activity of the LPS isolated from Acetobacterpasteurianus CIP103108. International Journal of Biological Macromolecules. 2018;119:1027-1035. doi: https://doi.org/10.1016/j.ijbiomac.2018.08.035
45. Moreira AP, Texeira TF, Ferreira AB, et al. Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br J Nutr. 2012;108(5):801-809. doi: https://doi.org/10.1017/S0007114512001213
46. Покусаева Д.П., Аниховская И.А., Коробкова Л.А. и др. Прогностическая значимость показателей системной эндотоксинемии в атерогенезе // Физиология человека. — 2019. — Т. 45. — №5. — С. 543-551.
47. Яковлев М.Ю. Роль кишечной микрофлоры и недостаточность барьерной функции печени в развитии эндотоксинемии и воспаления // Казанский медицинский журнал. — 1988. — Т. 69. — №5. — С. 353-358.
48. d’Hennezel E, Abubucker S, Murphy LO, Cullen TW. Total Lipopolysaccharide from the Human Gut Microbiome Silences Toll-Like Receptor Signaling. mSystems. 2017;2(6). doi: https://doi.org/10.1128/mSystems.00046-17
49. Popkin BM, Du S, Green WD, et al. Individuals with obesity and COVID-19: A global perspective on the epidemiology and biological relationships. Obes Rev. 2020;21(11):e13128. doi: https://doi.org/10.1111/obr.13128
50. Nagpal R, Newman TM, Wang S, et al. Obesity-Linked Gut Microbiome Dysbiosis Associated with Derangements in Gut Permeability and Intestinal Cellular Homeostasis Independent of Diet. J Diabetes Res. 2018;2018:1-9. doi: https://doi.org/10.1155/2018/3462092
51. Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761-1772. doi: https://doi.org/10.2337/db06-1491
52. Ciesielska A, Matyjek M, Kwiatkowska K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol Life Sci. 2021;78(4):1233-1261. doi: https://doi.org/10.1007/s00018-020-03656-y
53. Karpova T, de Oliveira AA, Naas H, et al. Blockade of Toll-like receptor 4 (TLR4) reduces oxidative stress and restores phospho-ERK1/2 levels in Leydig cells exposed to high glucose. Life Sciences. 2020;245:117365. doi: https://doi.org/10.1016/j.lfs.2020.117365
54. Li Y, Deng S-L, Lian Z-X, Yu K. Roles of Toll-Like Receptors in Nitroxidative Stress in Mammals. Cells. 2019; 8(6):576. doi: https://doi.org/10.3390/cells8060576
55. Frost RA, Nystrom GJ, Lang CH. Lipopolysaccharide regulates proinflammatory cytokine expression in mouse myoblasts and skeletal muscle. Am J Physiol Integr Comp Physiol. 2002;283(3):R698-R709. doi: https://doi.org/10.1152/ajpregu.00039.2002
56. Kong X, Yang Y, Ren L, et al. Activation of autophagy attenuates EtOH-LPS-induced hepatic steatosis and injury through MD2 associated TLR4 signaling. Sci Rep. 2017;7(1):9292. doi: https://doi.org/10.1038/s41598-017-09045-z
57. Song MJ, Kim KH, Yoon JM, Kim JB. Activation of toll-like receptor 4 is associated with insulin resistance in adipocytes. Biochemical and Biophysical Research Communications. 2006;346:739-745. doi: https://doi.org/10.1016/j.bbrc.2006.05.170
58. Okorokov PL, Anychovskaya IA, Yakovleva MM, et al. Nutritional factors of inflammation induction or lipid mechanism of endotoxin transport. Hum Physiol. 2012;38(6):649-655. doi: https://doi.org/10.1134/S0362119712060102
59. 59. Harte AL, Varma MC, Tripathi G, et al. High fat intake leads to acute postprandial exposure to circulating endotoxin in type 2 diabetic subjects. Diabetes Care. 2012;35:375-382. doi: https://doi.org/10.2337/dc11-1593
60. Lassenius MI, Pietilainen KH, Kaartinen K, et al. Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care. 2011;34:1809-1815. doi: https://doi.org/10.2337/dc10-2197
61. Monte SV, Caruana JA, Ghanim H, et al. Reduction in endotoxemia, oxidative and inflammatory stress, and insulin resistance after Roux-en-Y gastric bypass surgery in patients with morbid obesity and type 2 diabetes mellitus. Surgery. 2012;151:587-593. doi: https://doi.org/10.1016/j.surg.2011.09.038
62. Cox AJ, Zhang P, Bowden DW, et al. Increased intestinal permeability as a risk factor for type 2 diabetes. Diabetes and Metabolism. 2017;43:163-166. doi: https://doi.org/10.1016/j.diabet.2016.09.004
63. Harte AL, da Silva NF, Creely SJ, et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. Journal of Inflammation (London England). 2010;7:15. doi: https://doi.org/10.1186/1476-9255-7-15
64. Kallio KAE, Hätönen KA, Lehto M, et al. Endotoxemia, nutrition, and cardiometabolic disorders. Acta Diabetol. 2015;52(2):395-404. doi: https://doi.org/10.1007/s00592-014-0662-3
65. Усаченко Ю.В., Белоглазов В.А., Гордиенко А.И. Системное воспаление, уровень липополисахарид-связывающего белка и растворимых sСD14 рецепторов при коморбидности сезонного аллергического ринита, эссенциальной артериальной гипертензии и ожирения // Патогенез. — 2020. — Т. 18. — №3. — С. 61-67.
66. Liu X, Lu L, Yao P, et al. Lipopolysaccharide binding protein, obesity status and incidence of metabolic syndrome: A prospective study among middleaged and older chinese. Diabetologia. 2014;57:1834-1841. doi: https://doi.org/10.1007/s00125-014-3288-7
Белоглазов Владимир Алексеевич, д.м.н., профессор; Scopus Author ID: 7007129056; eLibrary SPIN: 7455-2188
Симферополь
Яцков Игорь Анатольевич; Scopus Author ID: 57218873902; eLibrary SPIN: 2395-5710
Россия, 295491, Симферополь, ул. Мальченко, д. 7
Кумельский Евгений Дмитриевич, аспирант; eLibrary SPIN: 7455-2188
Половинкина Валерия Владимировна, студент
Симферополь
|
|
1. Рисунок 1. Изменение баланса провоспалительных и противовоспалительных адипокинов и цитокинов при ожирении. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(135KB)
|
Метаданные ▾ | |
|
|
2. Рисунок 2. Основные пути поступления и метаболизма LPS. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(298KB)
|
Метаданные ▾ | |
Белоглазов В.А., Яцков И.А., Кумельский Е.Д., Половинкина В.В. Метаболическая эндотоксинемия: возможные причины и последствия. Ожирение и метаболизм. 2021;18(3):320-326. https://doi.org/10.14341/omet12750
Beloglazov V.A., Yatskov I.A., Kumelsky E.D., Polovinkina V.V. Metabolic endotoxemia: possible causes and consequences. Obesity and metabolism. 2021;18(3):320-326. (In Russ.) https://doi.org/10.14341/omet12750
 |
117292
Россия, Москва, ул. Дм. Ульянова, д.11
____________________________________
117292
11, Dm. Ul’yanova str., Moscow, Russian Federation



































