




Содержание
Перейти к:
https://doi.org/10.14341/omet12839
Перейти к:
Ожирение — хроническая патология, которую эксперты Всемирной организации здравоохранения расценивают как эпидемию, основываясь на высоких темпах ежегодного прироста доли населения с избыточной массой тела практически во всех странах мира. Ожирение является ведущей причиной формирования резистентности к инсулину тканей организма и возникновения сахарного диабета 2-го типа. Это заболевание чревато серьезными осложнениями: возникновением и усугублением сердечно-сосудистой патологии, неалкогольной жировой болезни печени, появлением некоторых видов злокачественных новообразований и дисфункций репродуктивной системы. Жировая ткань, скелетные мышцы и печень играют уникальные роли в поддержании метаболического гомеостаза организма. Эти отличия обусловлены тканеспецифичностью внутриклеточных сигнальных путей инсулина. В настоящем обзоре представлены данные литературы об особенностях молекулярных механизмов, ответственных за нарушение проведения регуляторных сигналов инсулина на внутриклеточном уровне в его главных органах-мишенях при ожирении. Изложены сведения о характере изменений межорганных метаболических потоков, обусловленных ростом массы жировой ткани, и их участии в формировании инсулинорезистентности печени и мышц. Обсуждается значимость дальнейшего углубленного изучения тканевых особенностей механизмов патогенеза инсулинорезистентности для разработки новых таргетных фармпрепаратов, которые послужат совершенствованию комплексной медикаментозной коррекции нарушений обмена у больных сахарным диабетом 2 типа.
Кузьменко Д.И., Климентьева Т.К., Самойлова Ю.Г., Денисов Н.С., Матвеева М.В. Особенности молекулярных механизмов патогенеза инсулинорезистентности в различных тканях при ожирении. Ожирение и метаболизм. 2022;19(4):410-417. https://doi.org/10.14341/omet12839
Kuzmenko D.I., Klimenteva T.K., Samoilova I.G., Denisov N.S., Matveeva M.V. Features of molecular mechanisms of insulin resistance pathogenesis in various tissues in obesity. Obesity and metabolism. 2022;19(4):410-417. (In Russ.) https://doi.org/10.14341/omet12839
Алиментарное ожирение приводит к формированию в организме больного метаболического синдрома (синдрома Х). Его признаками являются: висцеральное ожирение (рост массы жира брыжейки кишечника и сальников брюшной полости), повышение артериального давления, гиперхолестерол- и гипертриацилглицеролемии, а также гипергликемия натощак. Гипергликемия и гиперинсулинемия являются следствиями формирующейся системной инсулинорезистентности [1–4].Резистентность к инсулину можно определить как состояние организма больного, при котором в рамках физиологического диапазона флуктуаций концентрации инсулина в крови нарушен нормальный метаболический ответ всех органов-мишеней гормона: ослаблено подавление глюконеогенеза в печени и липолиза в жировой ткани, нарушается инсулинозависимый транспорт глюкозы в клетки и подавлен синтез гликогена в мышцах и печени. Пониженная чувствительность тканей к инсулину запускает компенсаторную активацию его секреции b-клетками островков Лангерганса поджелудочной железы. Повышенная нагрузка на железу приводит к декомпенсации b-клеток, что является обязательным компонентом патогенеза сахарного диабета 2 типа (СД2) [5]. На моделях СД2, вызванных диабетогенными рационами, а также при обследовании больных ожирением и СД2 с инсулинорезистентностью было установлено уменьшение количества рецепторов инсулина во всех типах его клеток-мишеней. Одной из причин этого феномена является активация убиквитинлигазы (membrane-associated ring-CH — type finger 1), с участием которой рецепторы удаляется с клеточной поверхности [6]. Кроме того, выявлен дефект функционирования самого рецептора, выражающийся в снижении активности тирозиновой протеинкиназы его цитоплазматических доменов [7]. Жировая ткань, скелетные мышцы и печень являются наиболее значимыми органами-мишенями для инсулина. В поддержании метаболического гомеостаза организма эти органы выполняют различные функции, что обусловлено тканеспецифичностью внутриклеточных сигнальных путей инсулина в каждом из них. Общепризнано, что в механизмах формирования резистентности к инсулину определяющую роль играют дефекты передачи сигнала инсулина на пострецепторном, то есть внутриклеточном уровне.
Алиментарное ожирение — результат дисбаланса между количеством энергии, поступающей в организм с пищей, и ее расходованием в процессе жизнедеятельности. Гипертрофия жировой ткани тесно взаимосвязана с формированием ее инсулинорезистентности [8]. Проявлениями дисфункций гипертрофирующейся жировой ткани являются, во-первых, формирующийся уже на ранних стадиях заболевания окислительный стресс, который затем охватывает скелетные мышцы и печень, во-вторых, множественные нарушения метаболизма и низкоинтенсивное воспаление. Эти явления взаимно усиливаются, формируя порочный круг [9].
Резистентность к инсулину адипоцитов выражается в существенном ослаблении инсулин-зависимого ингибирования липолиза депонированных триацилглицеролов (TAG). Дефицит сигнализации инсулином не позволяет в полной мере активировать фосфодиэстеразу циклического аденозинмонофосфата. В итоге сохраняются опосредованная протеинкиназой А активация липолитического каскада и фосфорилирование перилипина, которое обеспечивает сохранение «окон» в оболочке липидной капли и доступ субстрата к активному центру гормоночувствительной липазы адипоцитов [10]. В этих условиях адипоциты висцерального жира демонстрируют более активную липолитическую программу. Даже в норме они менее чувствительны кантилиполитическому действию инсулина, чем адипоциты подкожного жира [11]. Эти явления лежат в основе характерного для инсулинорезистентных адипоцитов метаболического маркера — повышенной концентрации неэстерифицированных жирных кислот (НЭЖК) в крови натощак. Показана прямая корреляция между выраженностью гиперлипидемии и степенью нарушения толерантности к глюкозе организма больных ожирением и СД2 [12][13]. Установлено, что у больных СД2 в жировой ткани подавлена экспрессия генов, кодирующих основной транспортер НЭЖК из крови в адипоциты: fatty acid binding protein 4. Следовательно, инсулинорезистентность жировой ткани не только «растормаживает» липолиз, но и нарушает транспорт НЭЖК вадипоциты [14]. Установлено, что перманентно активированный липолиз в инсулинорезистентной жировой ткани способствует формированию резистентности к инсулину печени. Так, увеличенная доставка НЭЖК из жировой ткани в печень стимулирует их окисление и повышение концентрации в гепатоцитах ацетил-КоА — аллостерического активатора пируваткарбоксилазы — ключевого фермента глюконеогенеза в печени [15]. Усиленный приток в печень НЭЖК, мобилизуемых в адипоцитах, способствует активации синтеза TAG и их накоплению в гепатоците. Таким образом, активированные субстрат-зависимые метаболические процессы, параллельно с непосредственными эффектами инсулина на печень, являются обязательными участниками формирования ее инсулинорезистентности [16].
В адипоцитах больных ожирением и СД2, как и в случае со скелетными мышцами и печенью, показано уменьшение количества рецепторов инсулина, сочетающееся с ингибированием активности их тирозиновой протеинкиназы [17]. В условиях роста массы жировой ткани происходит ее инфильтрация активированными макрофагами, усиленно продуцирующими воспалительные цитокины, прежде всего фактор некроза опухоли-a (TNF-α) и интерлейкин-6. Они выходят в кровоток и начинают действовать за пределами жировой ткани [18]. TNF-α является активатором семейства стресс-активируемых протеинкиназ, в частности, сериновой протеинкиназы, фосфорилирующей домен активации транскрипции в белке c-Jun: c-Jun N-terminal kinases (JNK) [19]. Твердо установлено, что в основе эффекта JNK лежит фосфорилирование Ser-307 в молекуле субстрата рецептора инсулина-1 (IRS-1). Это подавляет способность IRS-1 передавать сигнал инсулина «вниз по течению» [20][21]. Таким образом, инсулинрезистентная жировая ткань способна оказывать системное воздействие в первую очередь на печень за счет высвобождения НЭЖК и воспалительных цитокинов (рис. 1, B).
В норме до 30% глюкозы крови поступает в скелетные мышцы путем инсулин-стимулируемого транспорта. Масса скелетных мышц в организме взрослого человека составляет 35–42% массы тела. Мышцы не только обеспечивают существенный вклад в системную инсулинорезистентность, вызванную ожирением и СД2, но и влияют на метаболизм всего организма [22]. У лиц с инсулинорезистентностью скелетных мышц глюкоза крови по большей части перенаправляется в печень, где включается в липогенез. Вследствие активации синтеза TAG существенно повышается его внутриклеточная концентрация, что является основой эктопического накопления липидов в гепатоцитах и их жировой дистрофии [23]. Еще в 1994 г. на биоптатах скелетных мышц впервые было показано, что активность тирозиновой протеинкиназы рецепторов инсулина была снижена только у больных ожирением и СД2, но не изменялась у лиц с избыточной массой, без признаков инсулинорезистентности [24]. Прямой ингибирующий эффект на протеинкиназу рецептора может оказывать TNF-α, главным источником которого при ожирении становится увеличивающаяся в массе жировая ткань, гиперсекретирующая цитокин в кровяное русло, что позволяет TNF-α действовать за пределами жировой ткани [25].
Данные литературы свидетельствуют о том, что TAG, накапливающиеся в цитоплазме инсулинорезистентных мышц, вряд ли могут непосредственно нарушать работу внутриклеточных сигнальных путей инсулина. На эту роль могут претендовать, как полагают, по меньшей мере два интермедиата обмена липидов: 1,2-диацилглицерол (1,2-DAG) и церамиды [26]. На моделях ожирения и СД2 показано, что во всех органах-мишенях инсулина не только значительно повышена концентрация 1,2-DAG, но и выявлена четкая ассоциация между повышенным внутриклеточным содержанием 1,2-DAG и TAG в мышцах и печени и инсулинорезистентностью этих органов [26]. Как в эксперименте, так и при наблюдении за волонтерами, получавшими инфузии липидов, отмечены существенное увеличение в инсулинорезистентных мышцах содержания 1,2-DAG и активация изоформы-θ подсемейства новых протеинкиназ С (nPKCθ) [27][28]. Аналогичные результаты были получены при обследовании биоптатов инсулинорезистентных скелетных мышц больных СД2 [29]. Эти данные согласуются с тем, что, во-первых, 1,2-DAG необходим для активации только классических и новых подсемейств протеинкиназы С (cPKC и nPKC) [30] и, во-вторых, 1,2-DAG-связывающий домен nPKC имеет существенно большее сродство к 1,2-DAG чем у cPKC [31]. Активация nPKCθ блокирует сигнальный путь инсулина, прежде всего на уровне белка — субстрата инсулинового рецептора-1 (IRS-1). nPKCθ фосфорилируетв молекуле IRS-1 остаток Ser-1101 [28], что создает в молекуле IRS-1 препятствие для фосфорилирования по остаткам тирозина и этим нарушает способность IRS-1 активировать фосфатидилинозитол-3-киназу (PI3K) [32]. Помимо этого, в экспериментах с миотрубочками L6 показано, что nPKCθ способна фосфорилировать остатки Ser-504 и Ser-532 в составе фосфатидилинозитол фосфат-зависимой киназы-1 (PDK1), что приводило к нарушению PDK1-опосредованного фосфорилирования Tyr-308 в составе молекулы протеинкиназы В (PKB), необходимого для ее активации [33] (рис. 1, A).
Подобно инсулинорезистентным мышцам, в печени при ожирении и СД2 признаки формирования инсулинорезистентности гепатоцитов прослеживаются уже на уровне рецептора инсулина: уменьшается количество рецепторов и снижается активность рецепторной тирозиновой протеинкиназы [34]. У больных СД2 в инсулинорезистентной печени существенно увеличена активность глюконеогенеза, что является непосредственной причиной гипергликемии натощак — одного изметаболических маркеров заболевания [35]. Известно, что в норме инсулин способствует липогенезу в печени. Однако инсулинорезистентная печень демонстрирует парадоксальный ответ: липогенез перманентно активирован. Такая ситуация усугубляется «растормаживанием» липолиза TAG, депонированных в резистентной к инсулину жировой ткани. Это характерное проявление метаболических взаимодействий между жировой тканью и печенью в условиях ожирения и СД2 получило название косвенного эффекта инсулина на печень или «липолитического контроля» метаболизма печени. Освобождаемые в адипоцитах НЭЖК доставляются кровью в печень, обеспечивая синтез дополнительным количеством субстратов, что способствует возникновению неалкогольной жировой болезни печени [8]. Гены, кодирующие ключевые ферменты синтеза TAG de novo, находятся под контролем транскрипционных факторов ChREBP (carbohydrate-responsive element-binding protein) иSREBP-1 (sterol regulatory element-binding protein-1) [36]. Глюкоза — индуктор этих транскрипционных факторов. В условиях СД2 гипергликемия усиливает этот эффект [37]. Представленные данные подтверждают гипотезу, согласно которой избыточное накопление TAG в гепатоцитах является одним из важных звеньев патогенеза инсулинорезистентности печени [38].
Попытки идентифицировать изоформы PKC, участвующие в формировании инсулинорезистентности печени, выявили отличия от таковых в скелетных мышцах, где ведущую роль играет nPKCθ. На модели печеночной инсулинорезистентности, вызываемой трехдневной высокожировой диетой, изучали активности изоформ РКС, экспрессируемых в печени крыс. Показано, что увеличивалась активность только изоформы-e подсемейства nPKC (nPKCe) [39]. Эти результаты были подтверждены висследованиях как в эксперименте [40], так и при работе с биопсиями печени больных ожирением [41]. Во всех упомянутых исследованиях наблюдали прямую корреляцию между содержанием 1,2-DAG в гепатоцитах, активностью nPKCe иинсулинорезистентностью печени.
Было также показано, что nPKCe ответственна за ингибирование активности тирозиновой протеинкиназы рецептора инсулина, в результате фосфорилирования остатка Thr-1160 в составе пептидной петли этой протеинкиназы. Фосфорилирование Thr-1160 искажает нативную конформацию петли, что становится причиной ингибирования протеинкиназы рецептора [42]. Реальность этого механизма подтверждена исследованиями с использованием мутации Thr1160Glu, которая обеспечивала появление неактивной протеинкиназы рецептора. Другая мутация — Thr1160Ala — лишала способности протеинкиназы рецептора отвечать на ингибирующее действие nPKCe [42]. Также было показано, что мыши с нокаутом гена InsrT1160A оказались устойчивы к формированию инсулинорезистентности печени в ответ на кормление пищей с высоким содержанием жира [42]. Таким образом, в патогенезе инсулинорезистентности печени при ожирении и СД2 активирующаяся nPKCe не только угнетает активность киназы рецептора инсулина, но и затрудняет трансдукцию сигнала гормона на этапе IRS-1 🡒 PI3K (рис. 1, C).
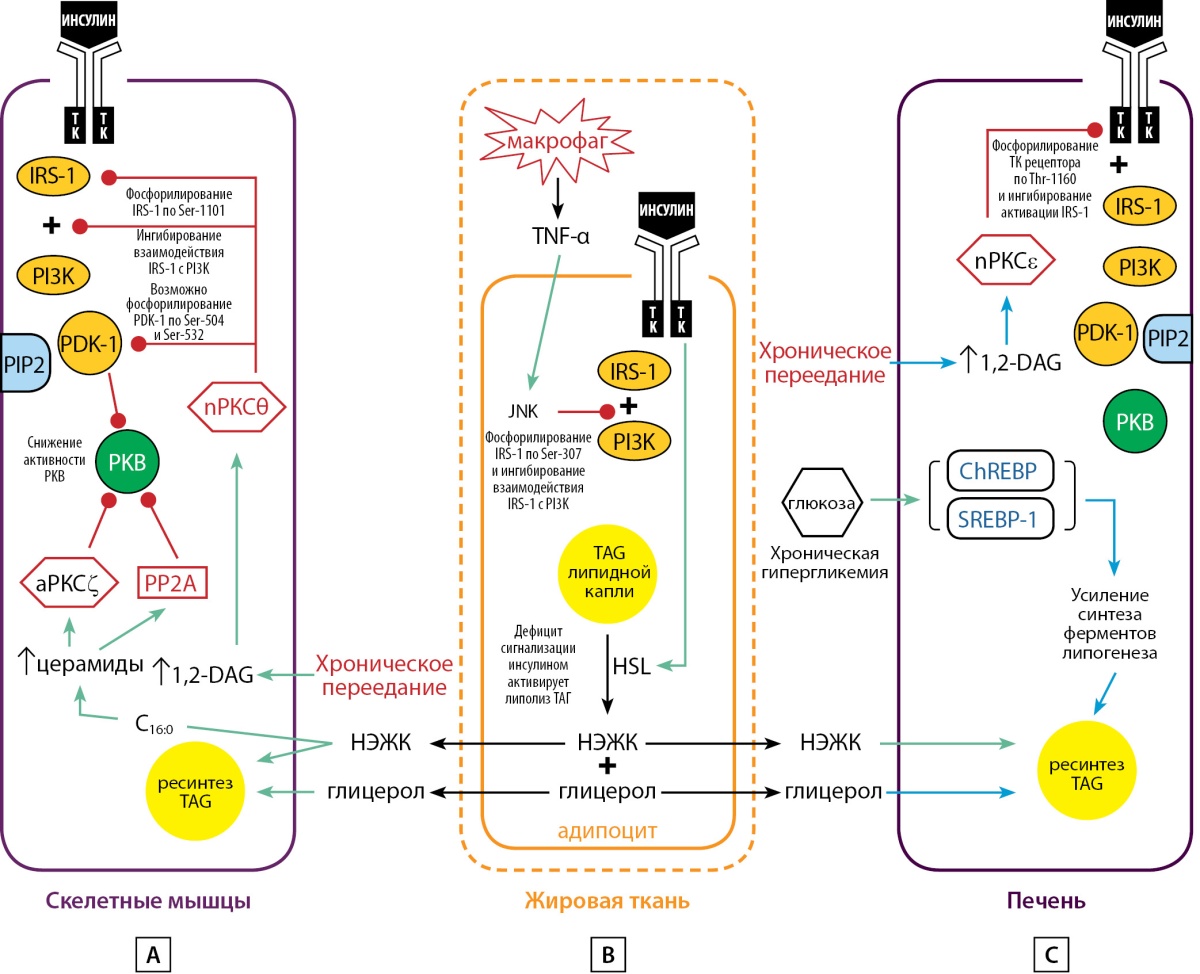
Рисунок 1. Предполагаемые ведущие молекулярные механизмы патогенеза инсулинорезистентности.
A — скелетные мышцы; B — жировая ткань; C — печень.
Примечание. Зеленые стрелки — активация; красные линии — ингибирование и ингибирующее фосфорилирование/дефосфорилирование. ТК — тирозиновая протеинкиназа рецепторов инсулина; PКВ — протеинкиназа В; 1,2-DAG — 1,2-диацилглицерол; TAG — триацилглицерол; TNF-α — фактор некроза опухоли-a; аPКСζ — ζ-изоформа подсемейства атипичных протеинкиназ С; nPКСθ — θ-изоформа подсемейства новых протеинкиназ С; nPКСε — ε-изоформа подсемейства новых протеинкиназ С; НЭЖК — неэстерифицированные жирные кислоты; IRS-1 —субстрат инсулинового рецептора-1; PI3K — фосфоинозитид-3-киназа; PDK-1 — фосфоинозитид-зависимая протеинкиназа-1; PIP2 — фосфоинозитид(4,5) бисфосфат; Ser — серин; Thr — треонин; С16:0 — пальмитиновая кислота; JNK — c-Jun N-terminal kinase; РР2А — фосфопротеинфосфатаза 2А; HSL — гормоночувствительная липаза; ChREBP — углевод-реагирующий элемент-связывающий белок; SREBP-1 — стерол-регулирующий элемент-связывающий белок-1.
Figure 1. Proposed leading molecular mechanisms of the pathogenesis of insulin resistance in various tissues in obesity.
A — skeletal muscles; B — adipose tissue; C — liver.
Green arrows — activation; red lines, inhibition and inhibitory phosphorylation/dephosphorylation. ТК — insulin receptor tyrosine kinase; PКВ — protein kinase В; 1,2-DAG — 1,2-diacylglycerol; TAG — triacylglycerol; TNF-a — tumor necrosis factor-a; аPКСζ — ζ-isoform of the subfamily of atypical protein kinases C; nPКСθ — θ-isoform of the subfamily of novel protein kinases C; nPКСε — ε-isoform of the subfamily of novel protein kinases C; НЭЖК — non-esterified fatty acids; IRS-1 — substrate for insulin receptor-1; PI3K — phosphoinositide-3-kinase; PDK-1 — phosphoinositide-dependent kinase-1; PIP2 — phosphoinositide(4,5) bisphosphate; Ser — serine; Thr — threonine; С16:0 — palmitic acid; JNK — c-Jun N-terminal kinase; РР2А — phosphoprotein phosphatase 2A; HSL — hormone-sensitive lipase; ChREBP — carbohydrate-responsive element-binding protein; SREBP-1 — sterol regulatory element-binding protein-1.
В течение последнего десятилетия получены важные данные, указывающие на участие церамидов в мультифакторном патогенезе инсулинорезистентности при ожирении и СД2. К настоящему времени наиболее подробно эти аспекты изучены для скелетных мышц [43–45]. В организме человека церамид (N-ацилсфингозин), как ключевой метаболит сфинголипидов, представлен обширным семейством молекул, насчитывающим более 150 разновидностей [46]. Церамид состоит из сфингозина («структурная основа» молекулы), к С-2 атому которого амидной связью присоединена вариабельная по длине ацильная цепь. Церамиды выполняют множественные функции в организме, в том числе регулируют активность ряда метаболических процессов. Длина ацильной цепи в составе индивидуального церамида определяет его биологическую активность [47]. Пул церамидов с различной длиной ацильных цепей, способных нарушать проведение сигнала инсулина, синтезируется de novo в клетках скелетных мышц и печени. Этому способствует избыточное поступление длинноцепочечных НЭЖК из инсулинорезистентной жировой ткани благодаря перманентно активированному липолизу в адипоцитах. Синтез церамидов протекает в четыре этапа. На третьем этапе функционирует церамидсинтаза (ЦерС), катализирующая присоединение ацила к аминогруппе сфинганина [48]. Известны шесть изоформ ЦерС (ЦерС1 — ЦерС6), которые обладают субстратной специфичностью относительно длины присоединяемых ацил-КоА. Следовательно, активация конкретных изоформ ЦерС определяет появление церамидов с индивидуальной длиной ацильной цепи [49].
В висцеральном жире лиц с избыточной массой тела, а также в жировой ткани и печени мышей с ожирением, вызванным липидной диетой, были обнаружены сходные сдвиги обмена сфинголипидов: усиление экспрессии ЦерС6 и повышенное содержание С16:0-церамида. Усиление экспрессии ЦерС6 позитивно коррелировало со степенью выраженности инсулинорезистентности. Нокаут гена, кодирующего ЦерС6 у мышей, сопровождался снижением тканевого содержания С14:0- и С16:0-церамидов. Эти животные оказались устойчивы к ожирению, индуцированному липидной диетой, у них с трудом развивалась инсулинорезистентность [50]. В печени мышей доминантной изоформой является ЦерС2, катализирующая синтез церамидов сочень длинной ацильной цепью: С22:0-, С24:0- и С24:1-церамидов [49]. Нокаут гена, кодирующего ЦерС2, приводил к снижению содержания в печени С24:0- и С24:1-церамидов без существенного влияния на содержание С16:0-церамида [51]. В недавней работе S. Raichur и соавт. (2019), осуществленной на мышах с нокаутом гена, кодирующего ЦерС6, была подтверждена роль С16:0-церамидов в патогенезе инсулинорезистентности печени, индуцированной ожирением [52]. При исследовании профиля церамидов подкожного жира тучных женщин (индекс массы тела (ИМТ) 30–40 кг/м2) с жировой дистрофией печени (14% липидов в печени) было установлено, что в ткани значимо преобладали С18:0-, С18:1-, С22:0-, и С24:1-церамиды по сравнению с пациентками с нормальным уровнем липидов в печени [44]. Другими авторами в абдоминальном подкожном жире лиц с ожирением (ИМТ>30 кг/м2) показано достоверное увеличение содержания церамидов: на 31% у женщин и на 34% у мужчин по сравнению с группой здоровых лиц (ИМТ< 25 кг/м2). В ткани было повышено содержание С14:0-, С16:0- и С24:0-церамидов [53].
Предполагают, что церамиды могут участвовать в формировании инсулинорезистентности клеток путем ингибирования PKB, которая является центральным медиатором множественных эффектов инсулина. Во-первых, церамиды являются прямыми активаторами фосфопротеинфосфатазы 2А (РР2А) [54]. Под действием РР2А в молекуле PKB дефосфорилируются остатки Ser-473 и Thr-308, которые необходимы для стабилизации активной конформации протеинкиназы [55]. Во-вторых, церамиды активируют изоформу-ζ подсемейства атипичных PKC (аPKCζ), которая фосфорилирует остатки Ser и Thr в составе РН-домена молекулы PKB [56]. По этой причине PKВ лишается способности связываться с комплексом фосфатидилинозитол-3,4,5-трифосфат (PIP3) — PDK1 на плазматической мембране, что необходимо для перехода PKB в активное состояние [57] (рис. 1, А).
Данные литературы указывают на то, что системная резистентность к инсулину, обусловленная ожирением, формируется как с помощью тканеавтономных, так и перекрестно-зависимых патогенетических механизмов. Инсулинорезистентность характеризуется уменьшением количества рецепторов и снижением активности их тирозиновой протеинкиназы, которые, по-видимому, не имеют заметной тканеспецифичности. Имеющиеся сведения позволяют полагать, что ведущую роль в формировании пониженной чувствительности клеток к инсулину играют дефекты передачи гормонального сигнала на пострецепторном, или внутриклеточном, этапе. Особенности эффектов инсулина, регулирующих метаболизм миоцитов, адипоцитови гепатоцитов, находят свое отражение в тканеспецифичности механизмов патогенеза инсулинорезистентности.
Для жировой ткани показана активация стресс-активируемой протеинкиназы JNK, которая фосфорилирует специфические остатки серина в цитозольных доменах рецептора инсулина и молекул IRS-1. В мышечных клетках выявлена активация подсемейства nPKCθ, в результате чего происходит торможение проведения сигнала инсулина на этапе IRS-1🡒PIP3🡒PDK1. В отличие от скелетных мышц, в инсулинорезистентных гепатоцитах ведущую роль, по-видимому, играет преимущественная активация подсемейства nPKCε, что приводит к ингибированию активности рецепторной тирозиновой протеинкиназы в результате фосфорилирования специфического остатка треонина в ее составе. Показано, что длинноцепочечные ацилцерамиды, накапливающиеся в клетках при алиментарном ожирении, способны активировать подсемейство aPKCζ и фосфопротеинфосфатазу РР2А. Изменение активностей этих ферментов затрудняет переход в активную форму PKB — главного медиатора сигналов инсулина внутри клетки. Получены веские аргументы в пользу того, что длинноцепочечные ацилцерамиды играют одну из ключевых ролей в механизмах нарушения проведения регуляторного сигнала инсулина на внутриклеточном уровне.
Детализация знаний об особенностях молекулярных механизмов патогенеза инсулинорезистентности различных тканей позволит выявить новые потенциальные молекулярные мишени для таргетных фармакологических препаратов. Они позволят оптимизировать комплексную медикаментозную коррекцию нарушенной чувствительности тканей к инсулину и обеспечат более надежную вторичную профилактику осложнений СД2.
Источники финансирования. Работа выполнена за счет средств грантов: Грант Президента МД3664-2022.3 и Грант РНФ 22-25-00632.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи.
Участие авторов: Кузьменко Д.И. — разработка концепции и дизайна статьи, поиск, анализ и интерпретация данных литературы, написание статьи, составление схемы; Климентьева Т.К. — поиск, анализ и интерпретация данных литературы, существенный вклад в концепцию статьи, внесение в рукопись существенной правки с целью повышения научной ценности статьи; Самойлова Ю.Г. — анализ и интерпретация данных литературы, внесение в рукопись существенной правки с целью повышения научной ценности статьи; Денисов Н.С. — поиск, анализ и интерпретация данных литературы, написание статьи; Матвеева М.В. — интерпретация данных литературы, существенный вклад в концепцию статьи.
Все авторы одобрили финальную версию статьи перед публикацией, выразили согласие нести ответственность за все аспекты работы, подразумевающую надлежащее изучение и решение вопросов, связанных с точностью или добросовестностью любой части работы.
1. Дедов И.И., Ткачук В.А., Гусев Н.Б., и др. Сахарный диабет 2 типа и метаболический синдром: молекулярные механизмы, ключевые сигнальные пути и определение биомишеней для новых лекарственных средств // Сахарный диабет. — 2018. — Т. 21. — №5. — С. 364-375. doi: https://doi.org/10.14341/DM9730
2. Saltiel AR. Insulin signaling in health and disease. J Clin Invest. 2021;131(1):e142241. doi: https://doi.org/10.1172/JCI142241
3. Sakurai Y, Kubota N, Toshimasa Y, Kadowaki T. Role of insulin resistance in MAFLD. Int J Mol Sci. 2021;22 (8):4156-4182. doi: https://doi.org/10.3390/ijms22084156
4. Wu H, Ballantyne ChM. Metabolic inflammation and insulin resistance in obesity. CircRes. 2020;126 (11):1549-1564. doi: https://doi.org/10.1161/CIRCRESAHA.119.315896]
5. Czech MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med. 2017;23(7):804-814. doi: https://doi.org/10.1038/nm.4350
6. Nagarajan A, Petersen MC, Nasiri AR, et al. MARCH1 regulates insulin sensitivity by controlling cell surface insulin receptor levels. Nat Commun. 2016;7:12639. doi: https://doi.org/10.1038/ncomms12639
7. Wali JA, Jarzebska N, Raubenheimer D, et al. Cardio-metabolic effects of high-fat diets and their underlying mechanisms — a narrative review. Nutrients. 2020;12(5):1505-1523. doi: https://doi.org/10.3390/nu12051505
8. Longo M, Zatterale F, Naderi J, et al. Adipose tissue dysfunction as determinant of obesity-associated metabolic complications. Int J Mol Sci. 2019;20(9):2358-2381. doi: https://doi.org/10.3390/ijms20092358
9. Кузьменко Д.И., Удинцев С.А., Климентьева Т.К., Серебров В.Ю. Окислительный стресс жировой ткани как первичное звено патогенеза резистентности к инсулину // Биомедицинская химия. — 2016. — Т. 62. — №1. — С. 14-21. doi: https://doi.org/10.1134/S1990750816030100
10. Yang A, Mottillo EP. Adipocyte lipolysis:from molecular mechanisms of regulation to disease and therapeutics. Biochem J. 2020;477(5):985-1008. doi: https://doi.org/10.1042/BCJ20190468
11. Luong Q, Huang J, Lee KY. Deciphering white adipose tissue heterogeneity. Biology. 2019;8(2):23-37. doi: https://doi.org/10.3390/biology8020023/
12. Gastaldelli A, Gaggini M, DeFronzo RA. Role of Adipose Tissue Insulin Resistance in the Natural History of Type 2 Diabetes:Results From the San Antonio Metabolism Study. Diabetes. 2017;66:815-822. doi: https://doi.org/10.2337/db16-1167
13. S0ndergaard E, Espinosa De Ycaza AE, Morgan-Bathke M, Jensen MD. How to Measure Adipose Tissue Insulin Sensitivity. J Clin Endocrinol Metab. 2017;102(4):1193-1199. doi: https://doi.org/10.1210/jc.2017-00047
14. Bodis K, Roden M. Energy metabolism of white adipose tissue and insulin resistance in humans. Eur J Clin Invest. 2018;48 (11):e13017. doi: https://doi.org/10.1111/eci.13017
15. Goedeke L, Bates J, Vatner DF, et al. Acetyl-CoA carboxylase inhibition reverses NAFLD and hepatic insulin resistance but promotes hypertriglyceridemia in rodents. Hepatology. 2018;68 (6):2197-2211. doi: https://doi.org/10.1002/hep.30097
16. Perry RJ, Kim T, Zhang XM, et al. Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver-targeted mitochondrial uncoupler. Cell Metab. 2013;18(5):740-748. doi: https://doi.org/10.1016/j.cmet.2013.10.004
17. Cignarelli A, Genchi VA, Perrini S, et al. Insulin and insulin receptors in adipose tissue development. Int J Mol Sci. 2019;20 (3):759-779. doi: https://doi.org/10.3390/ijms2003075
18. Kern L, Mittenbuhler MJ, Vesting AJ, et al. Obesity-induced TNFa and IL-6 signaling:The missing link between obesity and inflammation-driven liver and colorectal cancers. Cancers (Basel). 2018;11 (1):24-45. doi: https://doi.org/10.3390/cancers11010024
19. Feng J, Lu Sh, Ou B, et al. The role of JNk signaling pathway in obesity-driven insulin resistance. Diabetes Metab Syndr Obes. 2020;13:1399-1406. doi: https://doi.org/10.2147/DMSO.S236127. eCollection 2020
20. Aguirre V, Uchida T, Yenush L, et al. The c-Jun NH2-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser307. J. Biol. Chem. 2000;275(12):9047-9054. doi: https://doi.org/10.1074/jbc.275.12.9047
21. Copps KD, Hancer NJ, Opare-Ado Let al. Irs1 serine 307 promotes insulin sensitivity in mice. Cell Metab. 2010;119:84-92. doi: https://doi.org/10.1016/j.cmet.2009.11.003
22. Merz KE, Thurmond DC. Role of skeletal muscle in insulin resistance and glucose uptake. Compr Physiol. 2020;10(3):785-809. doi: https://doi.org/10.1002/cphy.c19002926
23. Gancheva S, Jelenik T, Alvarez-Hernandez E, Roden M. Interorgan Metabolic Crosstalk in Human Insulin Resistance. Physiol Rev. 2018;98(3):1371-1415. doi: https://doi.org/0.1152/physrev.00015.2017
24. Nolan JJ, Freidenberg G, Henry R, et al. Role of human skeletal muscle insulin receptor kinase in the in vivo insulin resistance of noninsulin-dependent diabetes mellitus and obesity. J Clin Endocrin Metab. 1994;78(2):471-477. doi: https://doi.org/10.1210/jcem.78.2.8106637
25. Haeusler RA, McGraw TE, Accili D. Biochemical and cellular properties of insulin receptor signalling. Nat Rev Mol Cell Biol. 2018;9(1):31-44. doi: https://doi.org/10.1038/nrm.2017.89. Epub 2017 Oct 4
26. Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev. 2018;98(4):2133-2223. doi: https://doi.org/10.1152/physrev.00063.2017
27. Yu C, Chen Y, Cline GW, et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002;277(52):50230-50236. doi: https://doi.org/10.1074/jbc.M200958200
28. Szendroedi J, Yoshimura T, Phielix E, et al. Role of diacylglycerol activation of PKCtheta in lipid-induced muscle insulin resistance in humans. Proc Natl Acad Sci USA. 2014;111(26):9597-9602. doi: https://doi.org/10.1073/pnas.1409229111
29. Perreault L, Newsom SA, Strauss A, et al. Intracellular localization of diacylglycerols and sphingolipids influences insulin sensitivity and mitochondrial function in human skeletal muscle. JCI Insight. 2018;3(3):e96805. doi: https://doi.org/10.1172/jci.insight.96805
30. Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev. 2008;88(4):1341-1378. doi: https://doi.org/10.1152/physrev.00034.2007
31. Dries DR, Gallegos LL, Newton AC. A single residue in the C1 domain sensitizes novel protein kinase C isoforms to cellular diacylglycerol production. J Biol Chem. 2007;282(2):826-830. doi: https://doi.org/10.1074/jbc.C600268200
32. Hanger NJ, Qiu W, Cherella C, et al. Insulin and metabolic stress stimulate multisite serine/threonine phosphorylation of insulin receptor substrate 1 and inhibit tyrosine phosphorylation. J Biol Chem. 2014;289(18):12467-12484. doi: https://doi.org/10.1074/jbc.M114.554162
33. Wang C, Liu M, Riojas RA, et al. Protein kinase C theta (PKCtheta)-dependent phosphorylation of PDK1 at Ser504 and Ser532 contributes to palmitate-induced insulin resistance. J Biol Chem. 2009;284(4):2038-2044. doi: https://doi.org/10.1074/jbc.M806336200
34. James DE, Stockli J, Birnbaum MJ. The aetiology and molecular landscape of insulin resistance. Nat Rev Mol Cell Biol. 2021;22(11):751-771. doi: https://doi.org/10.1038/s41580-021-00390-6. Epub 2021 Jul 20
35. Petersen MC, Vatner DF, Shulman GI. Regulation of hepatic glucose metabolism in health and disease. Nat Rev Endocrinol. 2017;13(10):572-587. doi: https://doi.org/10.1038/nrendo.2017.80. Epub 2017 Jul 21
36. S0ndergaard E, Nielsen S. VLDL triglyceride accumulation in skeletal muscle and adipose tissue in type 2 diabetes. Curr Opin Lipidol. 2018;29(1):42-47. doi: https://doi.org/10.1097/MOL.0000000000000471
37. Song Z, Yang H, Zhou L, Yang F. Glucose-Sensing Transcription Factor MondoA/ChREBP as Targets for Type 2 Diabetes:Opportunities and Challenges. Int J Mol Sci. 2019;20(20):E5132. doi: https://doi.org/10.3390/ijms20205132
38. Heeren J, Scheja L. Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol Metab. 2021:101238. doi: https://doi.org/10.1016/j.molmet.2021.101238. Epub 2021 Apr 20
39. Samuel VT, Liu Z-X, Qu X, et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279(31):32345-32353. doi: https://doi.org/10.1074/jbc.M313478200
40. Petersen MC, Shulman GI. Roles of Diacylglycerols and Ceramides in Hepatic Insulin Resistance. Trends Pharmacol Sci. 2017;38(7):649-665. doi: https://doi.org/10.1016/j.tips.2017.04.004
41. Kumashiro N, Erion DM, Zhang D, et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci USA. 2011;108(39):16381-16385. doi: https://doi.org/10.1073/pnas.1113359108
42. Petersen MC, Madiraju AK, Gassaway BM, et al. Insulin receptor Thr1160 phosphorylation mediates lipidinduced hepatic insulin resistance. J Clin Invest. 2016;126(11):4361-4371. doi: https://doi.org/10.1172/JCI86013
43. Mandal N, Grambergs R, Mondal K, et al. Role of ceramides in the pathogenesis of diabetes mellitus and its complications. J Diabetes Complications. 2021;35(2):107734. doi: https://doi.org/10.1016/j.jdiacomp.2020.107734
44. Sokolowska E, Blachnio-Zabielska A. The Role of ceramides in insulin resistance. Front Endocrinol (Lausanne). 2019;10:577. doi: https://doi.org/10.3389/fendo.2019.00577. eCollection 2019
45. Дылева Ю.А., Груздева О.В., Белик Е.В. Церамиды: фокус на ожирение // Ожирение и метаболизм. — 2020. — Т. 17. — №3. — С. 307-315. doi: https://doi.org/10.14341/omet12565
46. Bielawski J, Pierce JS, Snider J, et al. Sphingolipid analysis by high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS). Adv. Exp. Med. Biol. 2010;688:46-59. doi: https://doi.org/10.1007/978-1-4419-6741-1_3
47. Garic D, De Sanctis JB, Shah J, et al. Biochemistry of very-long-chain and long-chain ceramides in cystic fibrosis and other diseases: The importance of side chain. Prog Lipid Res. 2019;74:130-144. doi: https://doi.org/10.1016/j.plipres.2019.03.001. Epub 2019 Mar 12
48. Hannun YA, Obeid LM. Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol. 2018;19(3):175-191. doi: https://doi.org/10.1038/nrm.2017.107. Epub 2017 Nov 22
49. Stiban J, Tidhar R, Futerman AH. Ceramide synthases:roles in cell physiology and signaling. Adv Exp Med Biol. 2010;688:60-71. doi: https://doi.org/10.1007/978-1-4419-6741-1_4
50. Turpin SM, Nicholls HT, Willmes DM, et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 2014;20(4):678-686. doi: https://doi.org/10.1016/j.cmet.2014.08.002
51. Raichur S, Wang ST, Chan PW, et al. CerS2 haploinsufficiency inhibits b-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab. 2014;20(4):687-695. doi: https://doi.org/10.1016/j.cmet.2014.09.015
52. Raichur S, Brunner B, Bielohuby M, et al. The role of C16:0 ceramide in the development of obesity and type 2 diabetes:CerS6 inhibition as a novel therapeutic approach. Molecular Metabolism. 2019;21:36-50. doi: https://doi.org/10.1016/j.molmet.2018.12.008
53. Blachnio-Zabielska AU, Koutsari Ch, Tchkonia T, Jensen MD. Sphingolipid content of human adipose tissue:relationship to adiponectin and insulin resistance. Obesity (Silver Spring). 2012;20(12):2341-2347. doi: https://doi.org/10.1038/oby.2012.126
54. Teruel T, Hernandez R, Lorenzo M. Ceramide mediates insulin resistance by tumor necrosis factor-alpha in brown adipocytes by maintaining Akt in an inactive dephosphorylated state. Diabetes. 2001;50(11):2563-2571. doi: https://doi.org/10.2337/diabetes.50.11.2563
55. Schubert KM, Scheid MP, Duronio V. Ceramide inhibits protein kinase B/Akt by promoting dephosphorylation of serine 473. J. Biol. Chem. 2000; 275(18):13330-13335. doi: https://doi.org/10.1074/jbc.275.18.13330
56. Bourbon NA, Sandirasegarane L, Kester M. Ceramide-induced inhibition of Akt is mediated through protein kinase Czeta:implications for growth arrest. J. Biol. Chem. 2002; 277(5):3286-3292. doi: https://doi.org/10.1074/jbc.M110541200
57. Hajduch E, Turban S, Le Liepvre X, et al. Targeting of PKCzeta and PKB to caveolin-enriched microdomains represents a crucial step underpinning the disruption in PKB-directed signalling by ceramide. Biochem. J. 2008;410(2):369-379. doi: https://doi.org/10.1042/BJ20070936
Кузьменко Дмитрий Иванович – доктор медицинских наук, профессор кафедры биохимии и молекулярной биологии с курсом клинической лабораторной диагностики.
634050, Томск, ул. Московский тракт, д. 2
Researcher ID: J-5531-2013; Scopus Author ID: 6603341699
Нет
Климентьева Татьяна Константиновна – кандидат биологических наук.
Томск
Researcher ID: P-1269-2016; Scopus Author ID: 7801652730
Нет
Самойлова Юлия Геннадьевна - доктор медицинских наук, профессор.
Томск
Researcher ID: S-4436-2016; Scopus Author ID: 6603015302
Нет
Денисов Никита Сергеевич.
Томск
Нет
Матвеева Мария Владимировна - доктор медицинских наук, профессор.
Томск
Researcher ID: J-6338-2015; Scopus ID 57191909736
Нет
|
|
1. Рисунок 1. Предполагаемые ведущие молекулярные механизмы патогенеза инсулинорезистентности. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(567KB)
|
Метаданные ▾ | |
Кузьменко Д.И., Климентьева Т.К., Самойлова Ю.Г., Денисов Н.С., Матвеева М.В. Особенности молекулярных механизмов патогенеза инсулинорезистентности в различных тканях при ожирении. Ожирение и метаболизм. 2022;19(4):410-417. https://doi.org/10.14341/omet12839
Kuzmenko D.I., Klimenteva T.K., Samoilova I.G., Denisov N.S., Matveeva M.V. Features of molecular mechanisms of insulin resistance pathogenesis in various tissues in obesity. Obesity and metabolism. 2022;19(4):410-417. (In Russ.) https://doi.org/10.14341/omet12839
 |
117292
Россия, Москва, ул. Дм. Ульянова, д.11
____________________________________
117292
11, Dm. Ul’yanova str., Moscow, Russian Federation



































